We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
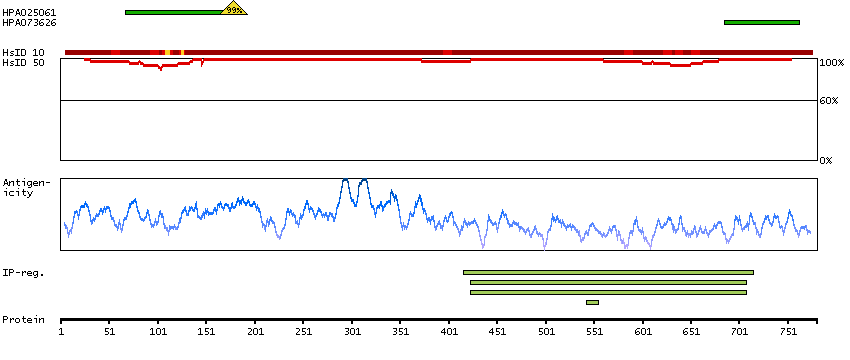
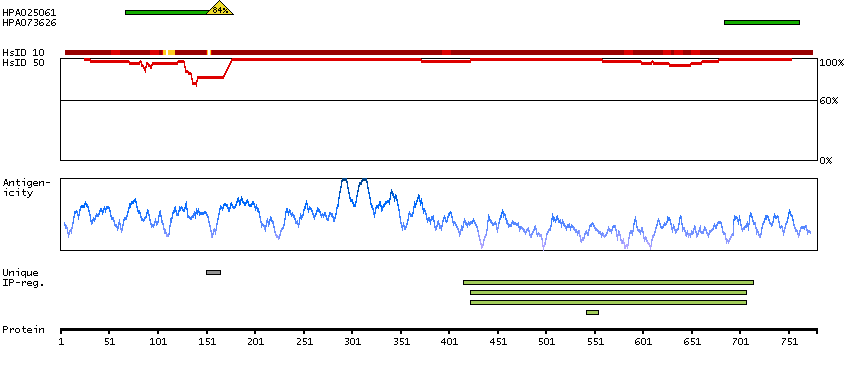
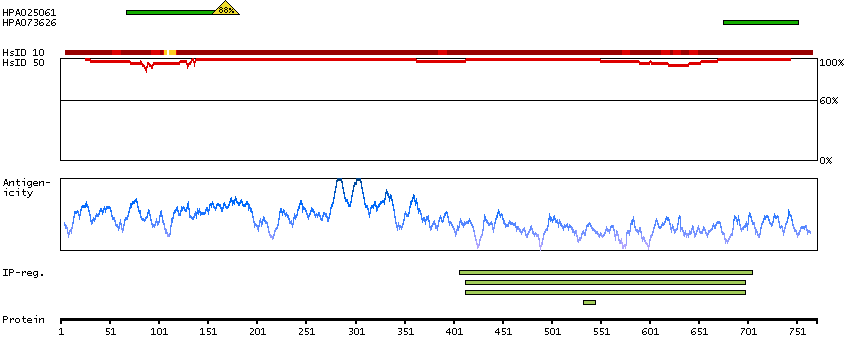
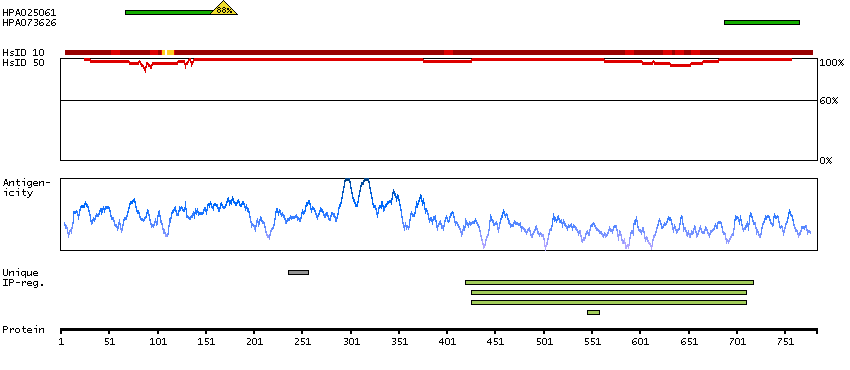
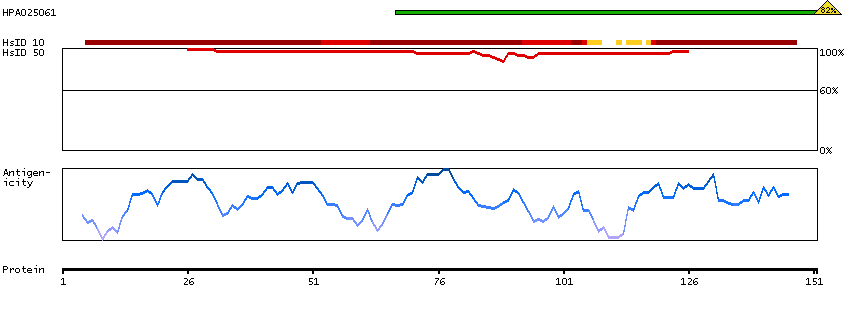
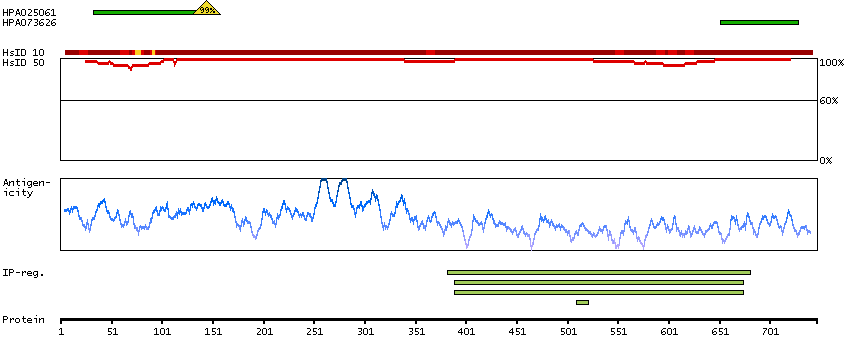
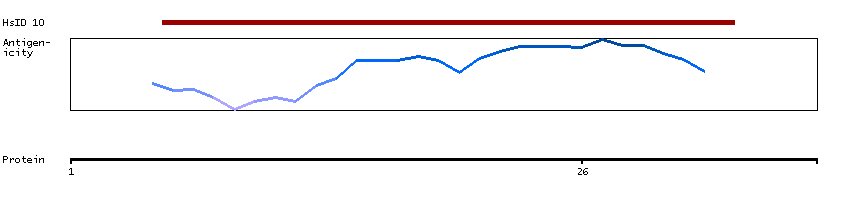
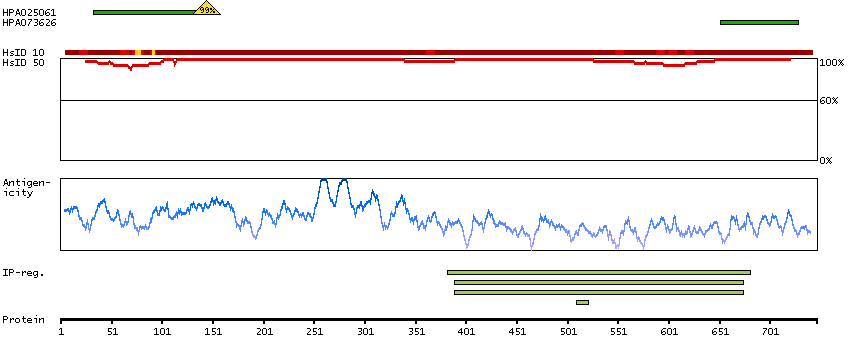
This gene encodes a member of the serine/threonine protein kinase family. Members of this kinase family are known to be essential for eukaryotic cell cycle control. Due to a segmental duplication, this gene shares very high sequence identity with a neighboring gene. These two genes are frequently deleted or altered in neuroblastoma. The protein kinase encoded by this gene can be cleaved by caspases and may play a role in cell apoptosis. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Sep 2015]