We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
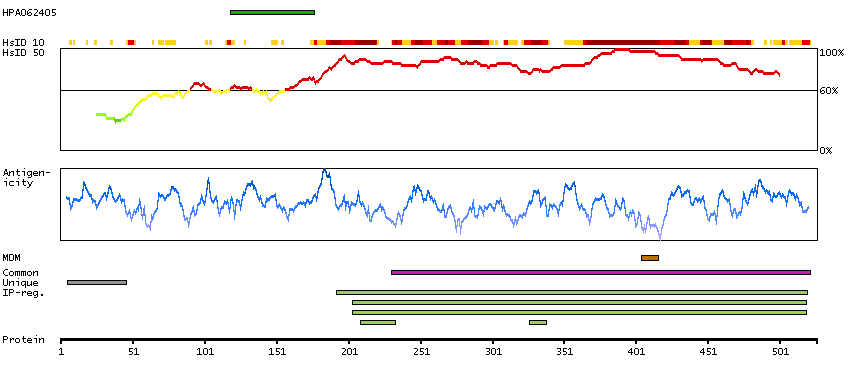
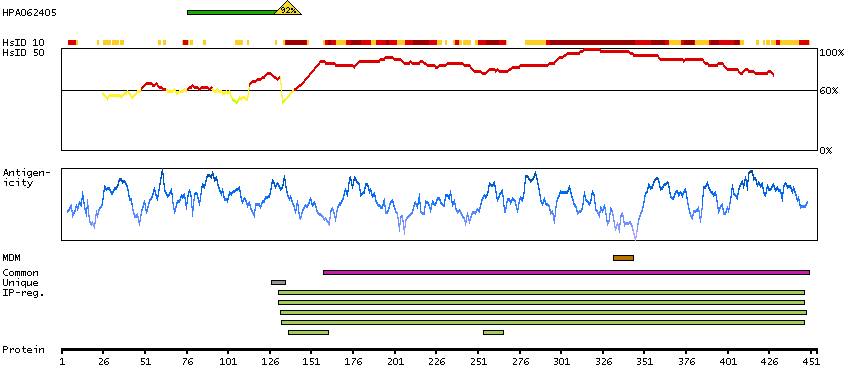
This gene encodes a member of the CDC2-like (or LAMMER) family of dual specificity protein kinases. In the nucleus, the encoded protein phosphorylates serine/arginine-rich proteins involved in pre-mRNA processing, releasing them into the nucleoplasm. The choice of splice sites during pre-mRNA processing may be regulated by the concentration of transacting factors, including serine/arginine rich proteins. Therefore, the encoded protein may play an indirect role in governing splice site selection. Multiple transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jun 2009]