We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
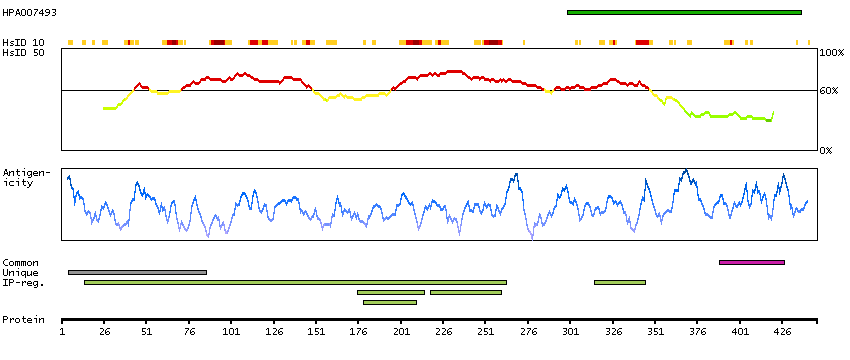
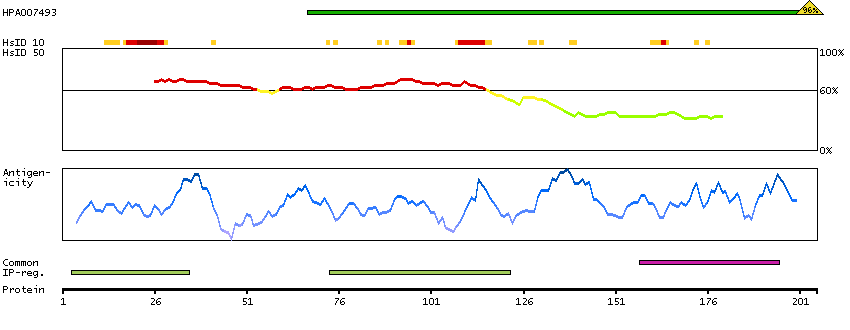
WD40 repeat proteins are key components of many essential biologic functions. They regulate the assembly of multiprotein complexes by presenting a beta-propeller platform for simultaneous and reversible protein-protein interactions. Members of the WIPI subfamily of WD40 repeat proteins, such as WIPI1, have a 7-bladed propeller structure and contain a conserved motif for interaction with phospholipids (Proikas-Cezanne et al., 2004 [PubMed 15602573]).[supplied by OMIM, Mar 2008]