We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
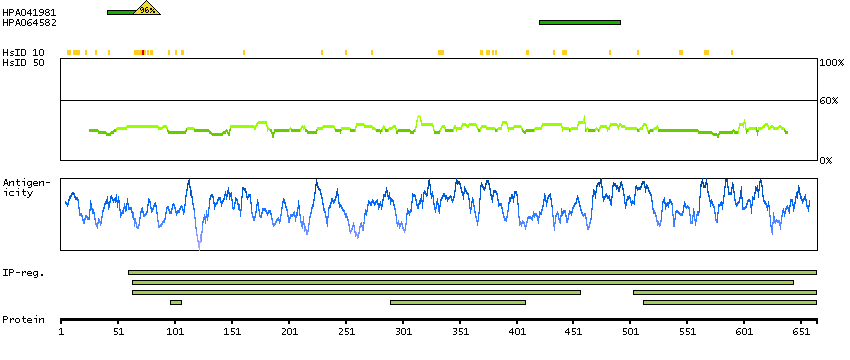
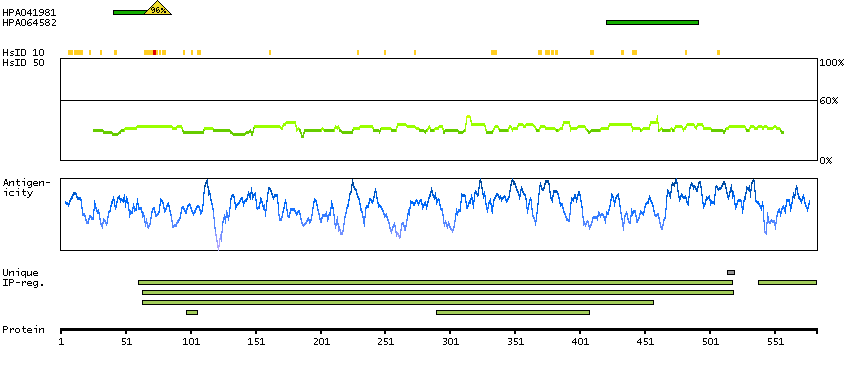
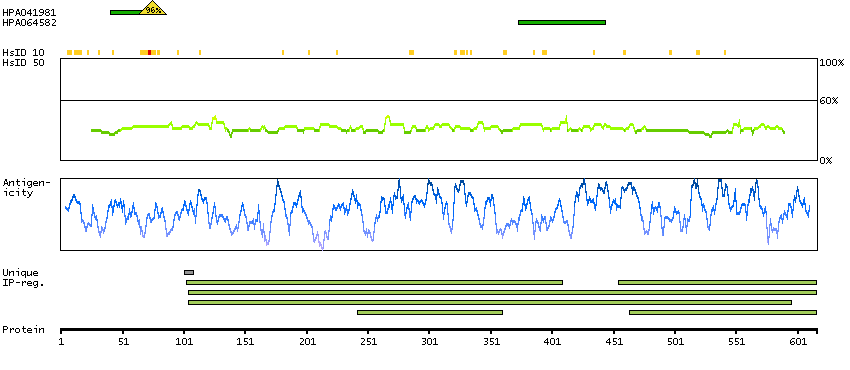
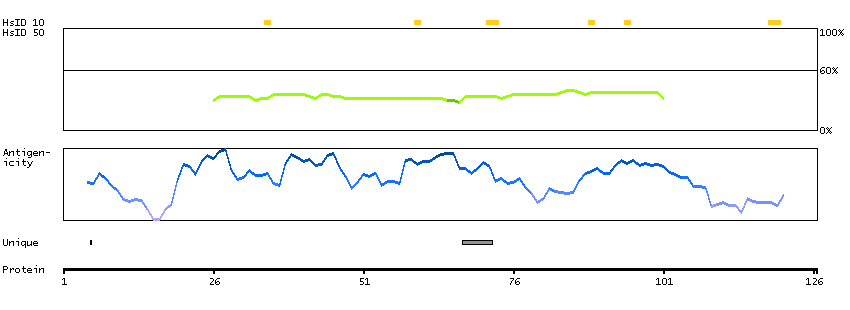
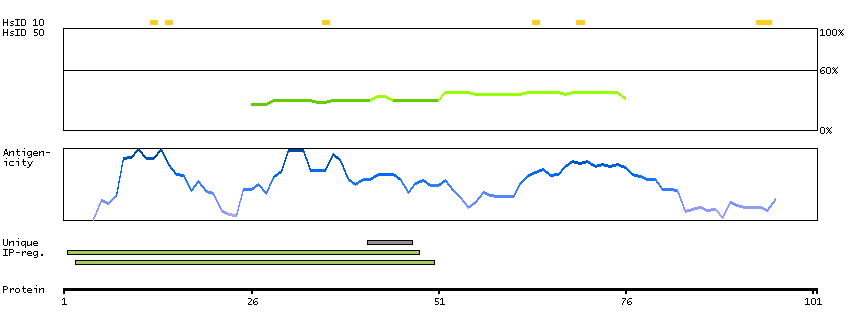
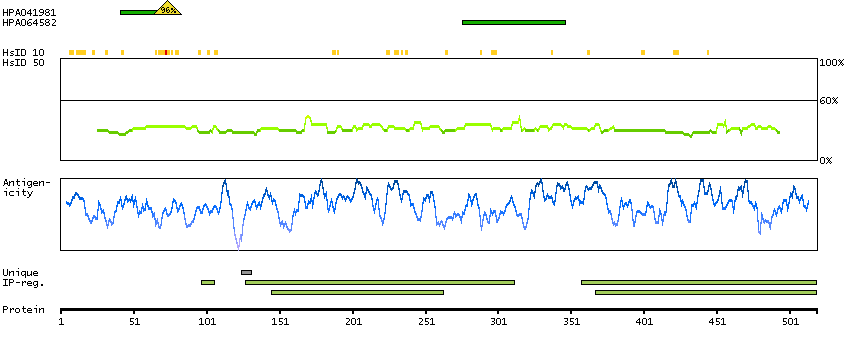
Succinate dehydrogenase complex, subunit A, flavoprotein (Fp) (HGNC Symbol)
Entrez gene summary
This gene encodes a major catalytic subunit of succinate-ubiquinone oxidoreductase, a complex of the mitochondrial respiratory chain. The complex is composed of four nuclear-encoded subunits and is localized in the mitochondrial inner membrane. Mutations in this gene have been associated with a form of mitochondrial respiratory chain deficiency known as Leigh Syndrome. A pseudogene has been identified on chromosome 3q29. Alternatively spliced transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jun 2014]
Enzymes ENZYME proteins Oxidoreductases SPOCTOPUS predicted membrane proteins Predicted intracellular proteins Plasma proteins Citric acid cycle related proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000104 [succinate dehydrogenase activity] GO:0005515 [protein binding] GO:0005739 [mitochondrion] GO:0005743 [mitochondrial inner membrane] GO:0005749 [mitochondrial respiratory chain complex II, succinate dehydrogenase complex (ubiquinone)] GO:0006099 [tricarboxylic acid cycle] GO:0006105 [succinate metabolic process] GO:0007399 [nervous system development] GO:0008177 [succinate dehydrogenase (ubiquinone) activity] GO:0016491 [oxidoreductase activity] GO:0016627 [oxidoreductase activity, acting on the CH-CH group of donors] GO:0022900 [electron transport chain] GO:0022904 [respiratory electron transport chain] GO:0043209 [myelin sheath] GO:0044237 [cellular metabolic process] GO:0044281 [small molecule metabolic process] GO:0050660 [flavin adenine dinucleotide binding] GO:0055114 [oxidation-reduction process]
Enzymes ENZYME proteins Oxidoreductases Predicted intracellular proteins Plasma proteins Citric acid cycle related proteins Disease related genes Potential drug targets Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000104 [succinate dehydrogenase activity] GO:0005515 [protein binding] GO:0005739 [mitochondrion] GO:0005743 [mitochondrial inner membrane] GO:0005749 [mitochondrial respiratory chain complex II, succinate dehydrogenase complex (ubiquinone)] GO:0006099 [tricarboxylic acid cycle] GO:0006105 [succinate metabolic process] GO:0007399 [nervous system development] GO:0008177 [succinate dehydrogenase (ubiquinone) activity] GO:0016491 [oxidoreductase activity] GO:0016627 [oxidoreductase activity, acting on the CH-CH group of donors] GO:0022900 [electron transport chain] GO:0022904 [respiratory electron transport chain] GO:0044237 [cellular metabolic process] GO:0044281 [small molecule metabolic process] GO:0050660 [flavin adenine dinucleotide binding] GO:0055114 [oxidation-reduction process]