We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (HGNC Symbol)
Entrez gene summary
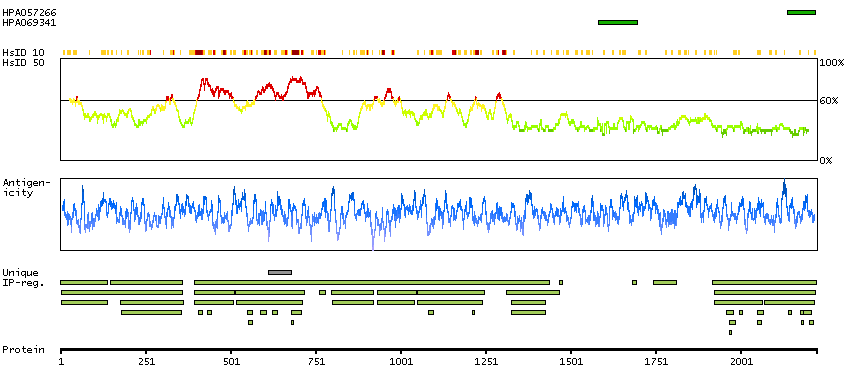
The de novo synthesis of pyrimidine nucleotides is required for mammalian cells to proliferate. This gene encodes a trifunctional protein which is associated with the enzymatic activities of the first 3 enzymes in the 6-step pathway of pyrimidine biosynthesis: carbamoylphosphate synthetase (CPS II), aspartate transcarbamoylase, and dihydroorotase. This protein is regulated by the mitogen-activated protein kinase (MAPK) cascade, which indicates a direct link between activation of the MAPK cascade and de novo biosynthesis of pyrimidine nucleotides. Alternative splicing results in multiple transcript variants encoding different isoforms. [provided by RefSeq, Apr 2015]
Enzymes ENZYME proteins Transferases Hydrolases Ligase SPOCTOPUS predicted membrane proteins Predicted intracellular proteins Plasma proteins Cancer-related genes Mutational cancer driver genes Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
SPOCTOPUS predicted membrane proteins Predicted intracellular proteins Cancer-related genes Mutational cancer driver genes Protein evidence (Ezkurdia et al 2014)