We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
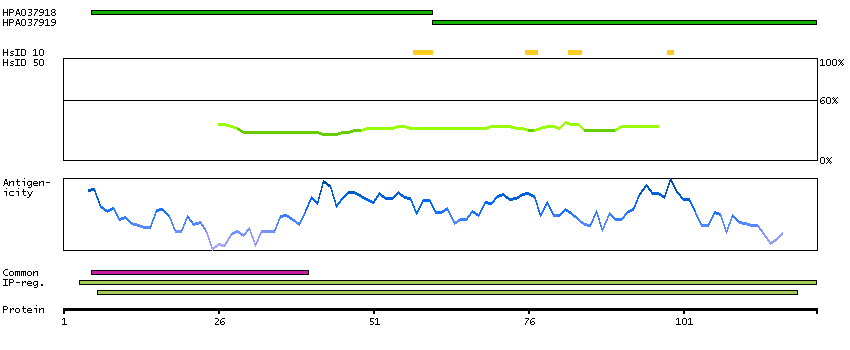
MEMSAT-SVM predicted membrane proteins Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0043066 [negative regulation of apoptotic process] GO:0045600 [positive regulation of fat cell differentiation] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter]
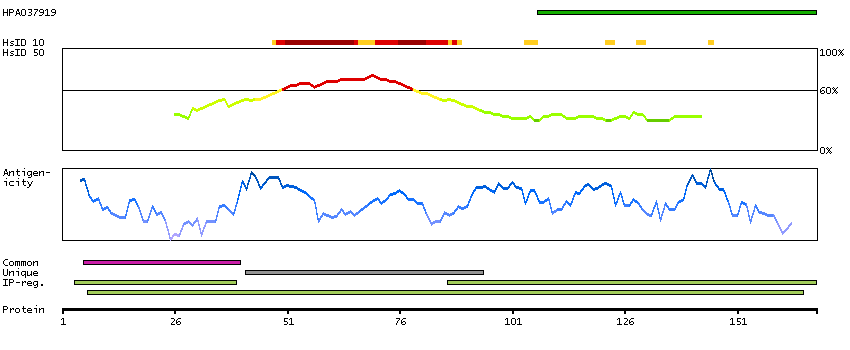
E9PR47 [Direct mapping] Mth938 domain-containing protein
Show all
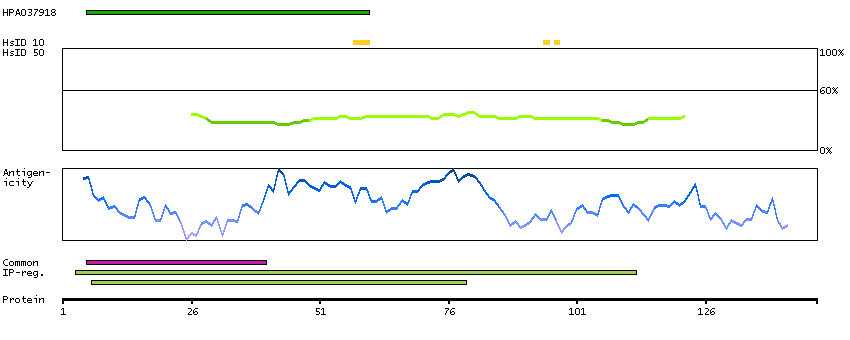
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0043066 [negative regulation of apoptotic process] GO:0045600 [positive regulation of fat cell differentiation] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter]
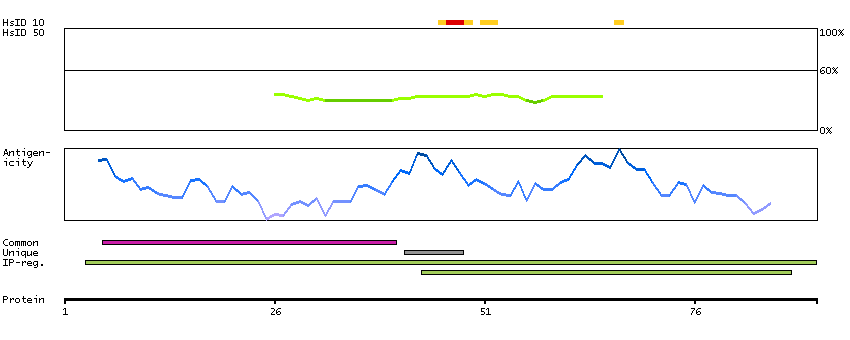
E9PIQ4 [Direct mapping] Mth938 domain-containing protein
Show all
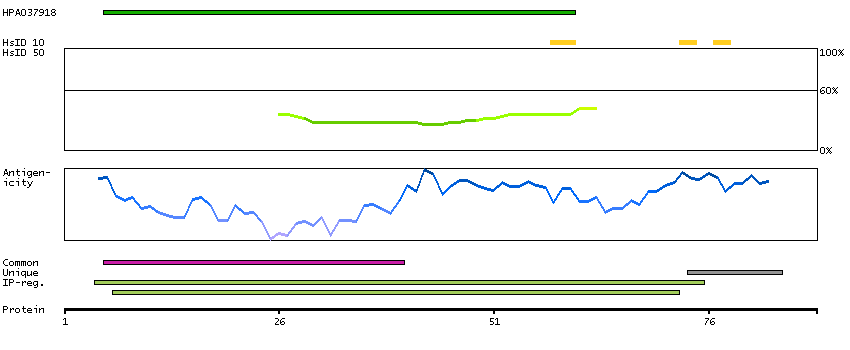
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0043066 [negative regulation of apoptotic process] GO:0045600 [positive regulation of fat cell differentiation] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter]
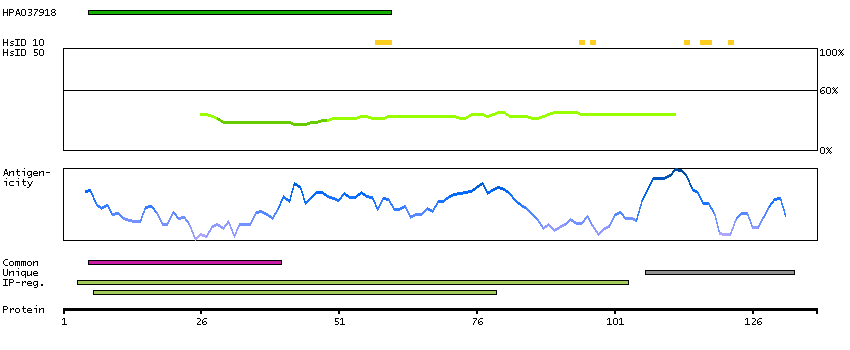
E9PLK9 [Direct mapping] Mth938 domain-containing protein
Show all
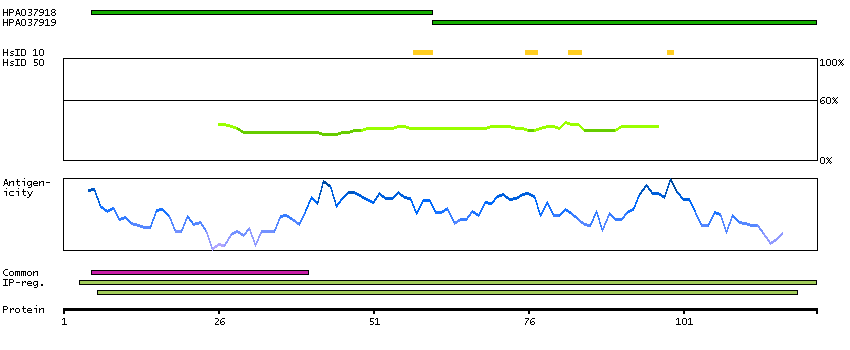
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0043066 [negative regulation of apoptotic process] GO:0045600 [positive regulation of fat cell differentiation] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter]
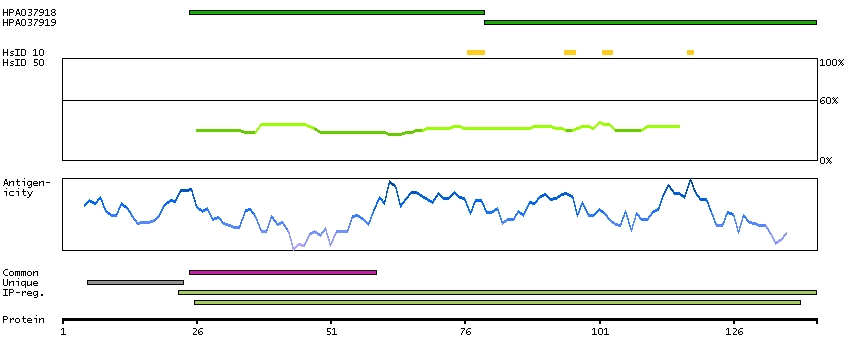
E9PNP3 [Direct mapping] Mth938 domain-containing protein
Show all
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0043066 [negative regulation of apoptotic process] GO:0045600 [positive regulation of fat cell differentiation] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter]
MEMSAT-SVM predicted membrane proteins Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0043066 [negative regulation of apoptotic process] GO:0045600 [positive regulation of fat cell differentiation] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter]