We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
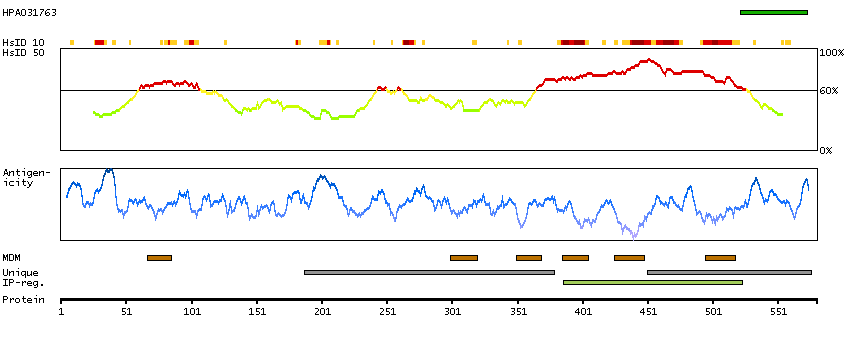
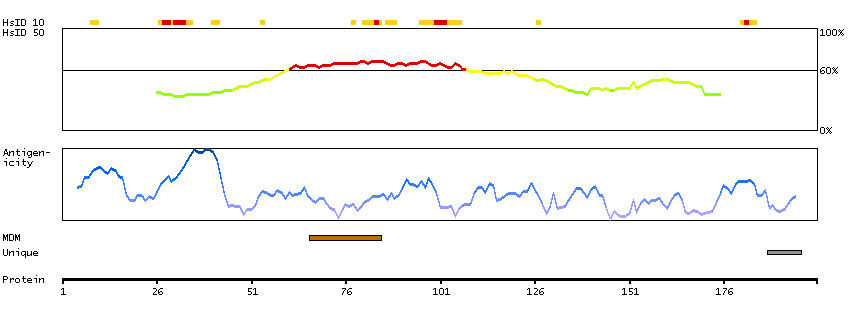
This gene encodes a memberof the transient receptor potential (TRP) cation channel gene family. The transmembrane protein localizes to intracellular vesicular membranes including lysosomes, and functions in the late endocytic pathway and in the regulation of lysosomal exocytosis. The channel is permeable to Ca(2+), Fe(2+), Na(+), K(+), and H(+), and is modulated by changes in Ca(2+) concentration. Mutations in this gene result in mucolipidosis type IV. [provided by RefSeq, Oct 2009]