We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
The protein encoded by this gene shares a significant similarity with Schizosaccharomyces pombe cdc5 gene product, which is a cell cycle regulator important for G2/M transition. This protein has been demonstrated to act as a positive regulator of cell cycle G2/M progression. It was also found to be an essential component of a non-snRNA spliceosome, which contains at least five additional protein factors and is required for the second catalytic step of pre-mRNA splicing. [provided by RefSeq, Jul 2008]
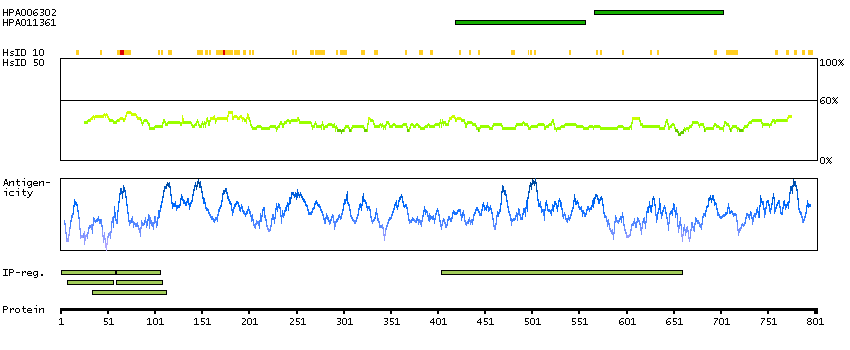
Q99459 [Direct mapping] Cell division cycle 5-like protein
Show all
Predicted intracellular proteins Plasma proteins Transcription factors Helix-turn-helix domains Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)