We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
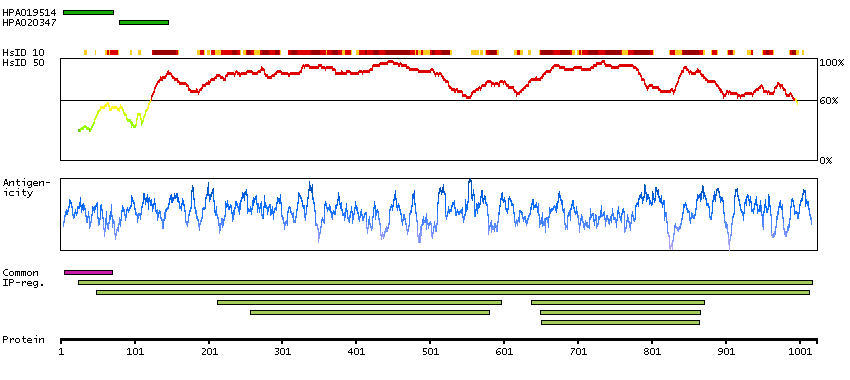
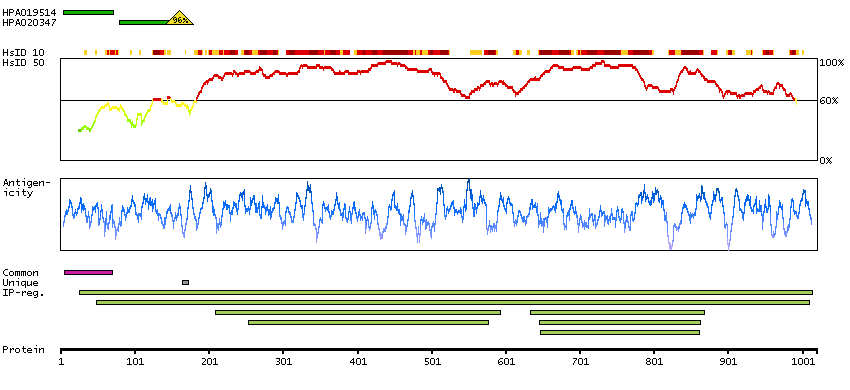
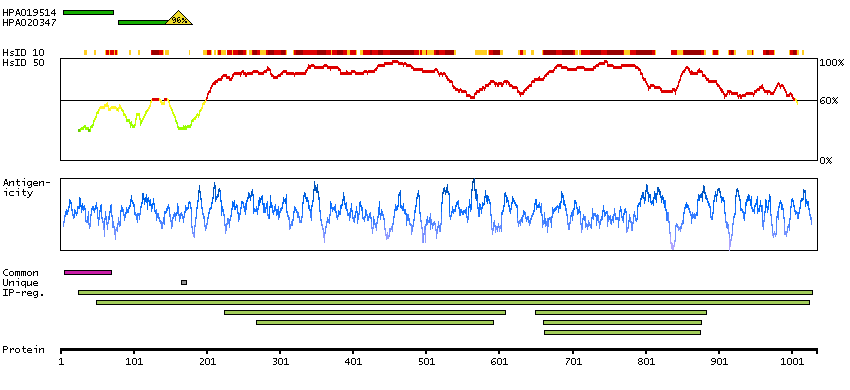
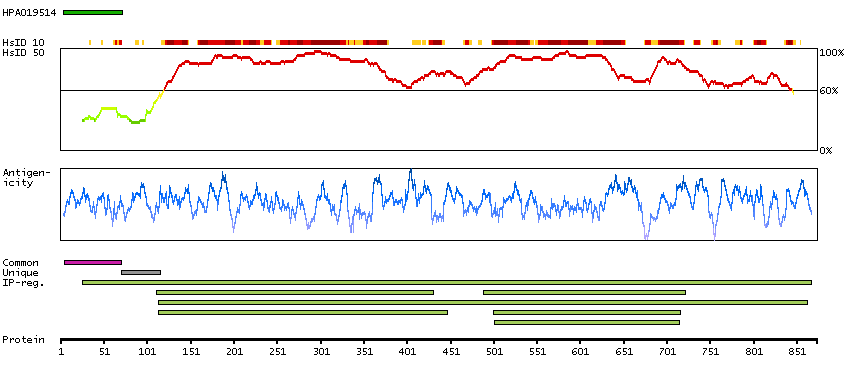
This gene encodes one subunit of the 2-oxoglutarate dehydrogenase complex. This complex catalyzes the overall conversion of 2-oxoglutarate (alpha-ketoglutarate) to succinyl-CoA and CO(2) during the Krebs cycle. The protein is located in the mitochondrial matrix and uses thiamine pyrophosphate as a cofactor. A congenital deficiency in 2-oxoglutarate dehydrogenase activity is believed to lead to hypotonia, metabolic acidosis, and hyperlactatemia. Alternative splicing results in multiple transcript variants encoding distinct isoforms.[provided by RefSeq, Sep 2009]
Enzymes ENZYME proteins Oxidoreductases Predicted intracellular proteins Plasma proteins Citric acid cycle related proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0004591 [oxoglutarate dehydrogenase (succinyl-transferring) activity] GO:0005739 [mitochondrion] GO:0005759 [mitochondrial matrix] GO:0006091 [generation of precursor metabolites and energy] GO:0006096 [glycolytic process] GO:0006099 [tricarboxylic acid cycle] GO:0006103 [2-oxoglutarate metabolic process] GO:0006104 [succinyl-CoA metabolic process] GO:0006554 [lysine catabolic process] GO:0006734 [NADH metabolic process] GO:0008152 [metabolic process] GO:0016624 [oxidoreductase activity, acting on the aldehyde or oxo group of donors, disulfide as acceptor] GO:0021695 [cerebellar cortex development] GO:0021756 [striatum development] GO:0021766 [hippocampus development] GO:0021794 [thalamus development] GO:0021860 [pyramidal neuron development] GO:0022028 [tangential migration from the subventricular zone to the olfactory bulb] GO:0030976 [thiamine pyrophosphate binding] GO:0031072 [heat shock protein binding] GO:0031966 [mitochondrial membrane] GO:0034602 [oxoglutarate dehydrogenase (NAD+) activity] GO:0034641 [cellular nitrogen compound metabolic process] GO:0044237 [cellular metabolic process] GO:0044281 [small molecule metabolic process] GO:0045252 [oxoglutarate dehydrogenase complex] GO:0046872 [metal ion binding] GO:0051087 [chaperone binding] GO:0055114 [oxidation-reduction process] GO:0061034 [olfactory bulb mitral cell layer development]
Enzymes ENZYME proteins Oxidoreductases Predicted intracellular proteins Plasma proteins Citric acid cycle related proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0004591 [oxoglutarate dehydrogenase (succinyl-transferring) activity] GO:0005739 [mitochondrion] GO:0005759 [mitochondrial matrix] GO:0006091 [generation of precursor metabolites and energy] GO:0006096 [glycolytic process] GO:0006099 [tricarboxylic acid cycle] GO:0006554 [lysine catabolic process] GO:0008152 [metabolic process] GO:0016624 [oxidoreductase activity, acting on the aldehyde or oxo group of donors, disulfide as acceptor] GO:0030976 [thiamine pyrophosphate binding] GO:0031966 [mitochondrial membrane] GO:0034641 [cellular nitrogen compound metabolic process] GO:0044237 [cellular metabolic process] GO:0044281 [small molecule metabolic process] GO:0045252 [oxoglutarate dehydrogenase complex] GO:0046872 [metal ion binding] GO:0055114 [oxidation-reduction process]
Enzymes ENZYME proteins Oxidoreductases Predicted intracellular proteins Plasma proteins Citric acid cycle related proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0004591 [oxoglutarate dehydrogenase (succinyl-transferring) activity] GO:0005739 [mitochondrion] GO:0005759 [mitochondrial matrix] GO:0006091 [generation of precursor metabolites and energy] GO:0006096 [glycolytic process] GO:0006099 [tricarboxylic acid cycle] GO:0006554 [lysine catabolic process] GO:0008152 [metabolic process] GO:0016624 [oxidoreductase activity, acting on the aldehyde or oxo group of donors, disulfide as acceptor] GO:0030976 [thiamine pyrophosphate binding] GO:0031966 [mitochondrial membrane] GO:0034641 [cellular nitrogen compound metabolic process] GO:0044237 [cellular metabolic process] GO:0044281 [small molecule metabolic process] GO:0045252 [oxoglutarate dehydrogenase complex] GO:0046872 [metal ion binding]