We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
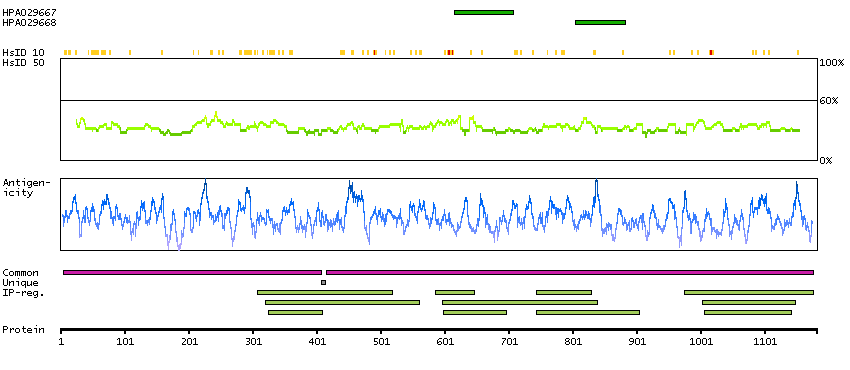
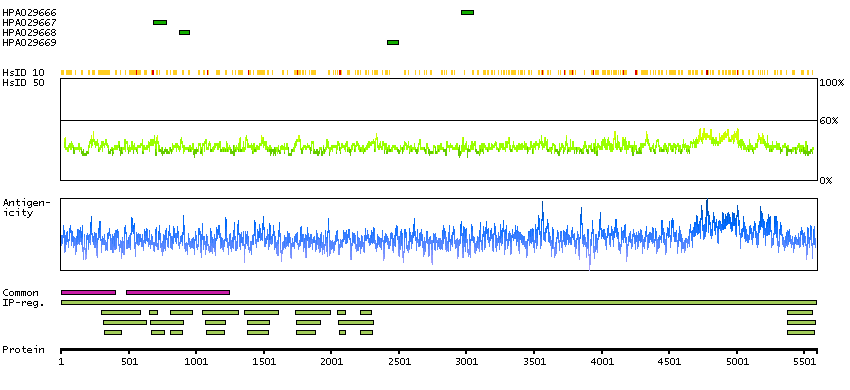
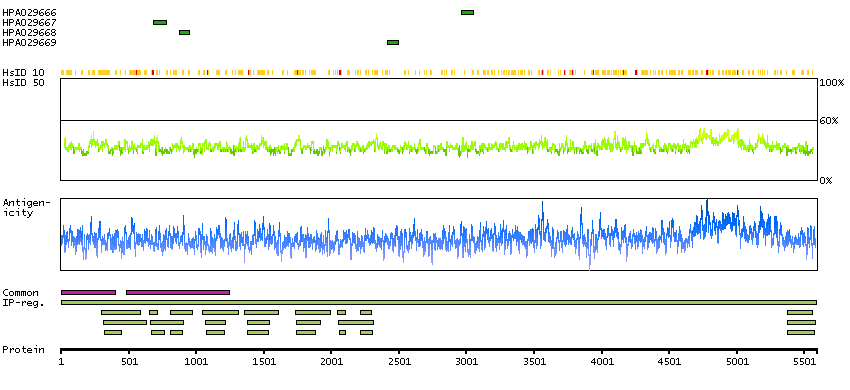
SPOCTOPUS predicted membrane proteins THUMBUP predicted membrane proteins Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)