We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
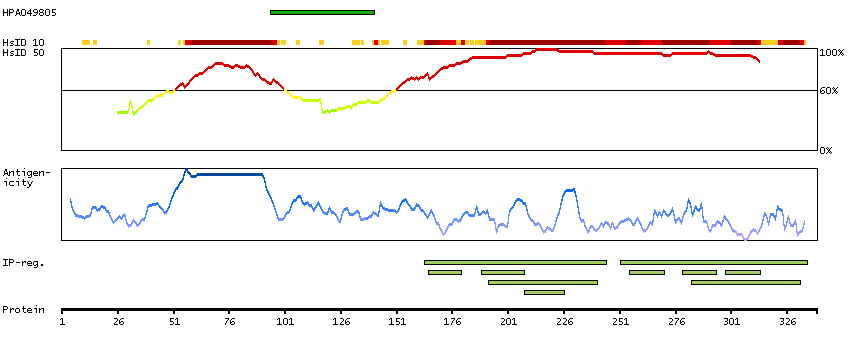
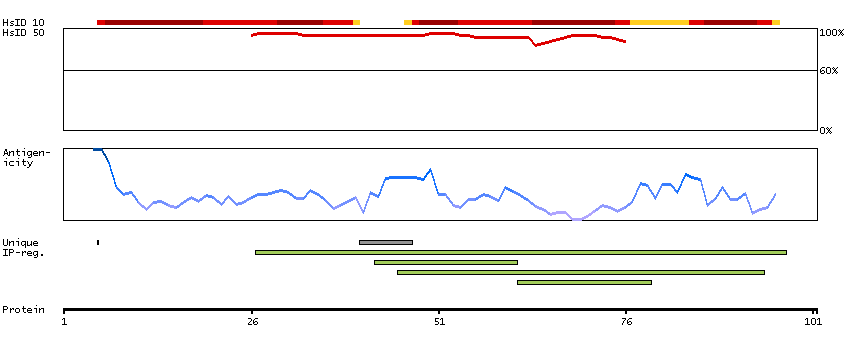
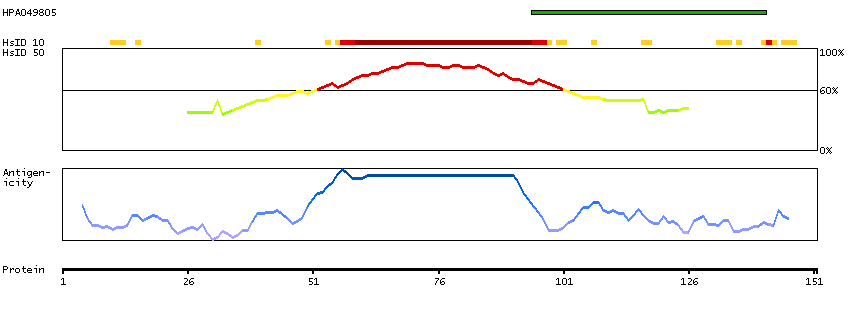
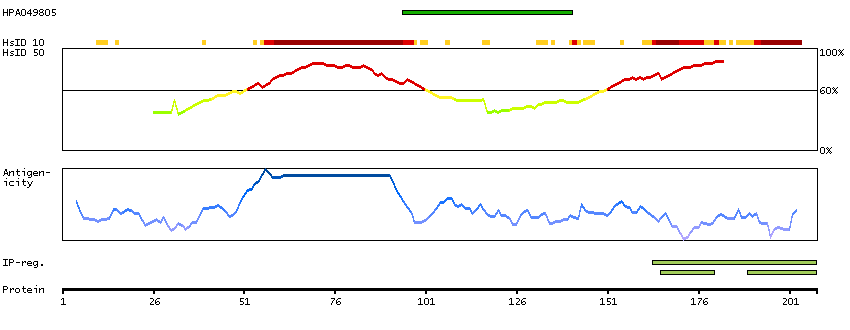
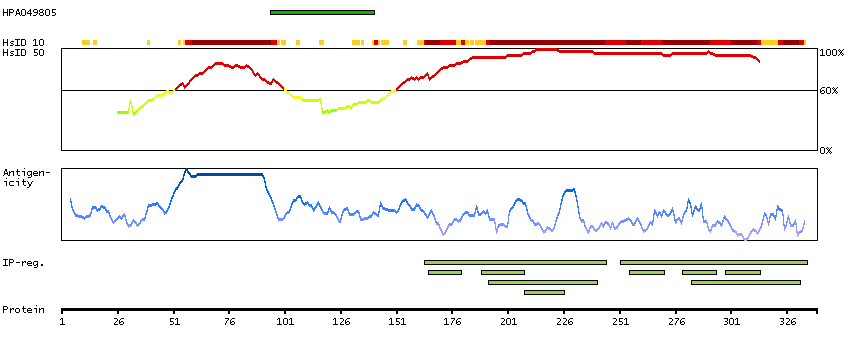
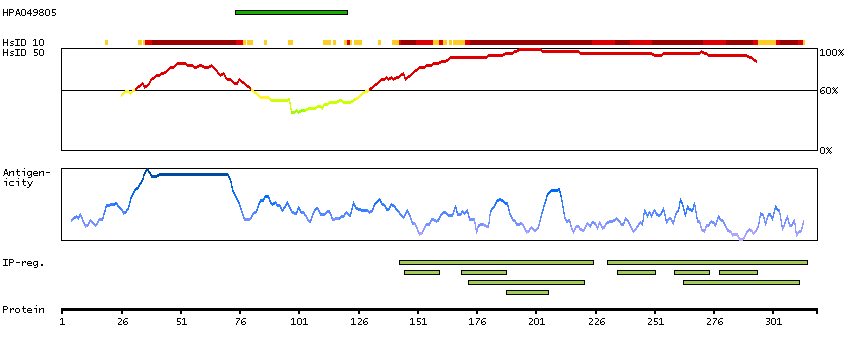
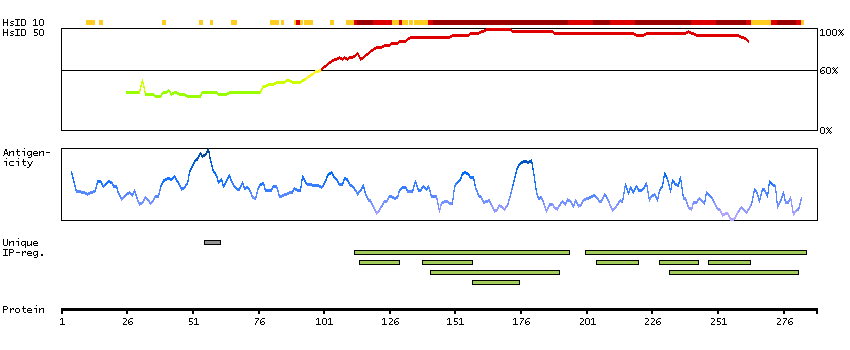
Initiation of transcription by RNA polymerase II requires the activities of more than 70 polypeptides. The protein that coordinates these activities is transcription factor IID (TFIID), which binds to the core promoter to position the polymerase properly, serves as the scaffold for assembly of the remainder of the transcription complex, and acts as a channel for regulatory signals. TFIID is composed of the TATA-binding protein (TBP) and a group of evolutionarily conserved proteins known as TBP-associated factors or TAFs. TAFs may participate in basal transcription, serve as coactivators, function in promoter recognition or modify general transcription factors (GTFs) to facilitate complex assembly and transcription initiation. This gene encodes TBP, the TATA-binding protein. A distinctive feature of TBP is a long string of glutamines in the N-terminus. This region of the protein modulates the DNA binding activity of the C terminus, and modulation of DNA binding affects the rate of transcription complex formation and initiation of transcription. The number of CAG repeats encoding the polyglutamine tract is usually 32-39, and expansion of the number of repeats increases the length of the polyglutamine string and is associated with spinocerebellar ataxia 17, a neurodegenerative disorder classified as a polyglutamine disease. Two transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Feb 2010]
Predicted intracellular proteins Transcription factors beta-Sheet binding to DNA Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000183 [chromatin silencing at rDNA] GO:0000978 [RNA polymerase II core promoter proximal region sequence-specific DNA binding] GO:0000979 [RNA polymerase II core promoter sequence-specific DNA binding] GO:0001103 [RNA polymerase II repressing transcription factor binding] GO:0001939 [female pronucleus] GO:0001940 [male pronucleus] GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005669 [transcription factor TFIID complex] GO:0005672 [transcription factor TFIIA complex] GO:0005719 [nuclear euchromatin] GO:0005737 [cytoplasm] GO:0006352 [DNA-templated transcription, initiation] GO:0006360 [transcription from RNA polymerase I promoter] GO:0006361 [transcription initiation from RNA polymerase I promoter] GO:0006362 [transcription elongation from RNA polymerase I promoter] GO:0006363 [termination of RNA polymerase I transcription] GO:0006366 [transcription from RNA polymerase II promoter] GO:0006367 [transcription initiation from RNA polymerase II promoter] GO:0006368 [transcription elongation from RNA polymerase II promoter] GO:0006383 [transcription from RNA polymerase III promoter] GO:0007283 [spermatogenesis] GO:0008134 [transcription factor binding] GO:0010467 [gene expression] GO:0016032 [viral process] GO:0019899 [enzyme binding] GO:0040029 [regulation of gene expression, epigenetic] GO:0044212 [transcription regulatory region DNA binding] GO:0045120 [pronucleus] GO:0045814 [negative regulation of gene expression, epigenetic] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0070491 [repressing transcription factor binding]
Predicted intracellular proteins Transcription factors beta-Sheet binding to DNA Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000183 [chromatin silencing at rDNA] GO:0003677 [DNA binding] GO:0005515 [protein binding] GO:0005654 [nucleoplasm] GO:0005669 [transcription factor TFIID complex] GO:0005672 [transcription factor TFIIA complex] GO:0005719 [nuclear euchromatin] GO:0006352 [DNA-templated transcription, initiation] GO:0006360 [transcription from RNA polymerase I promoter] GO:0006361 [transcription initiation from RNA polymerase I promoter] GO:0006362 [transcription elongation from RNA polymerase I promoter] GO:0006363 [termination of RNA polymerase I transcription] GO:0006366 [transcription from RNA polymerase II promoter] GO:0006367 [transcription initiation from RNA polymerase II promoter] GO:0006368 [transcription elongation from RNA polymerase II promoter] GO:0006383 [transcription from RNA polymerase III promoter] GO:0008134 [transcription factor binding] GO:0010467 [gene expression] GO:0016032 [viral process] GO:0019899 [enzyme binding] GO:0040029 [regulation of gene expression, epigenetic] GO:0044212 [transcription regulatory region DNA binding] GO:0045814 [negative regulation of gene expression, epigenetic] GO:0070491 [repressing transcription factor binding]