We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
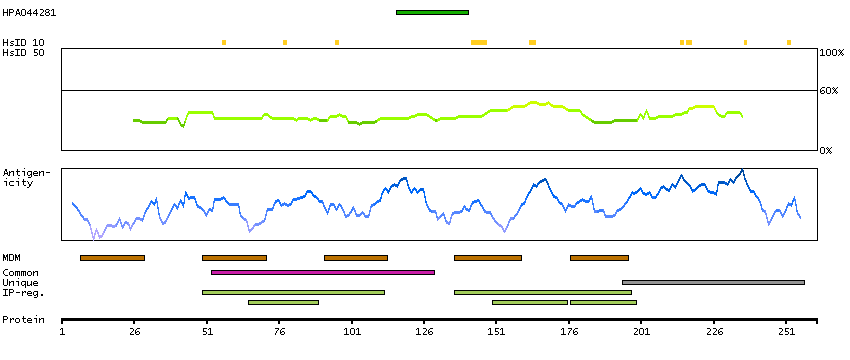
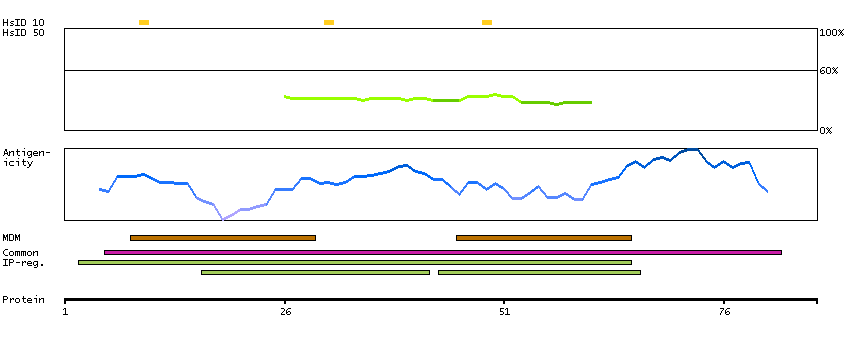
ATPase, H+ transporting, lysosomal 21kDa, V0 subunit b (HGNC Symbol)
Entrez gene summary
This gene encodes a portion of the V0 domain of vacuolar ATPase (V-ATPase), a multisubunit enzyme that mediates acidification of eukaryotic intracellular organelles. Activity of this enzyme is necessary for such varied processes as protein sorting, zymogen activation, receptor-mediated endocytosis, and synaptic vesicle proton gradient generation. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jun 2014]