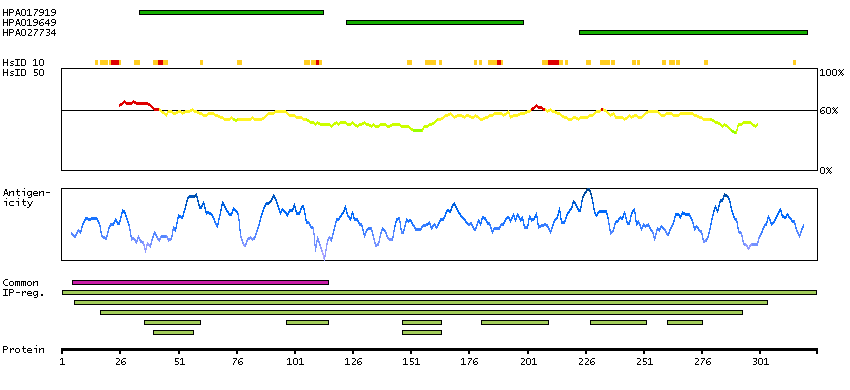
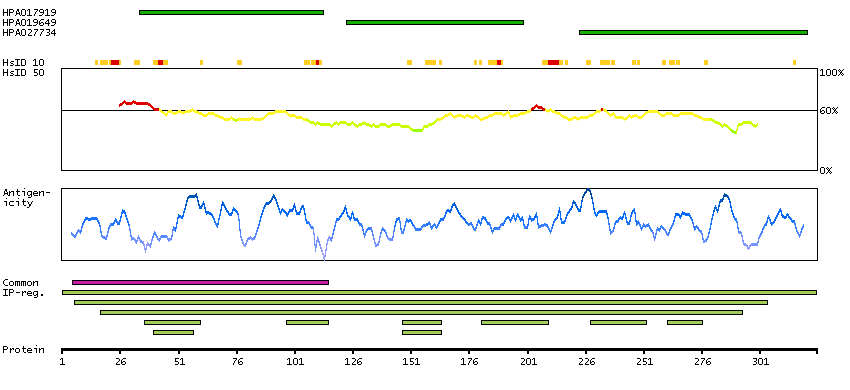
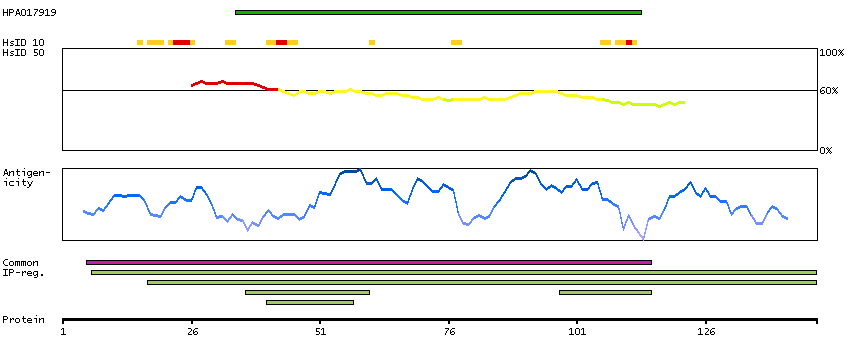
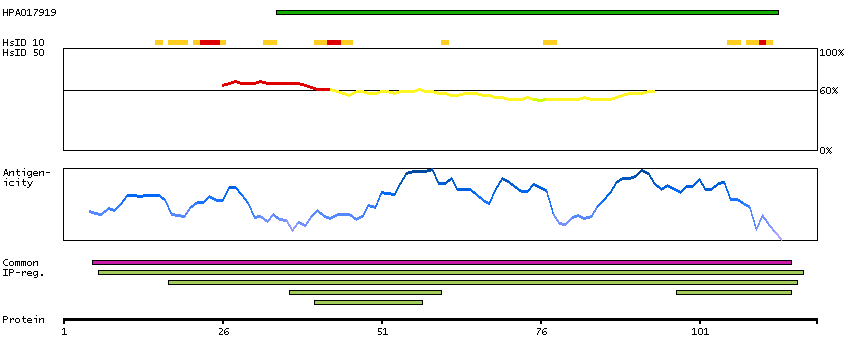
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
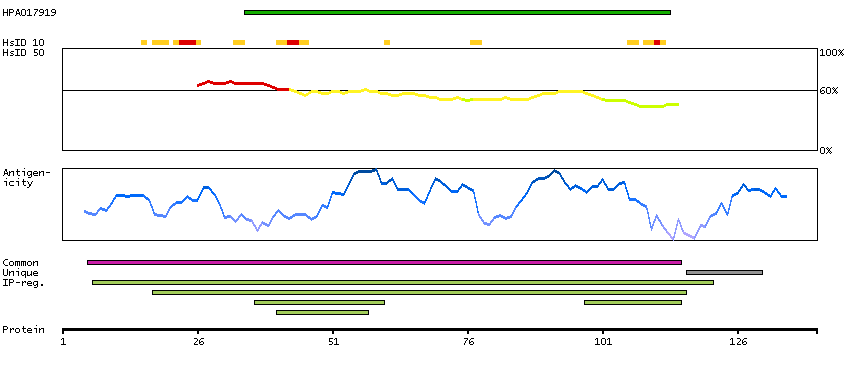
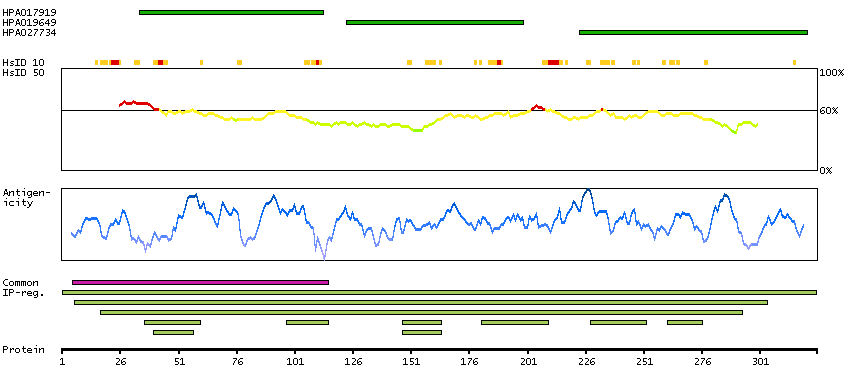
Aldo-keto reductase family 1, member A1 (aldehyde reductase) (HGNC Symbol)
Entrez gene summary
This gene encodes a member of the aldo/keto reductase superfamily, which consists of more than 40 known enzymes and proteins. This member, also known as aldehyde reductase, is involved in the reduction of biogenic and xenobiotic aldehydes and is present in virtually every tissue. Multiple alternatively spliced transcript variants of this gene exist, all encoding the same protein. [provided by RefSeq, Jan 2011]
P14550 [Direct mapping] Alcohol dehydrogenase [NADP(+)] V9HWI0 [Target identity:100%; Query identity:100%] Epididymis secretory protein Li 6; Epididymis secretory sperm binding protein Li 165mP
Show all
Enzymes ENZYME proteins Oxidoreductases Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
P14550 [Direct mapping] Alcohol dehydrogenase [NADP(+)] V9HWI0 [Target identity:100%; Query identity:100%] Epididymis secretory protein Li 6; Epididymis secretory sperm binding protein Li 165mP
Show all
Enzymes ENZYME proteins Oxidoreductases Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)