We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
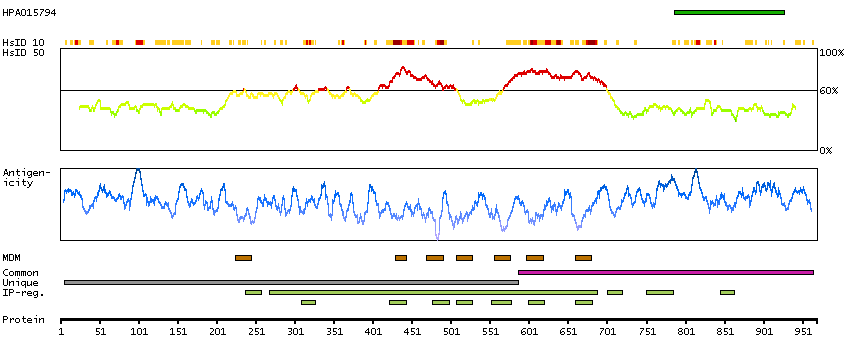
This gene encodes a member of the polycystin protein family. The encoded protein is a multi-pass membrane protein that functions as a calcium permeable cation channel, and is involved in calcium transport and calcium signaling in renal epithelial cells. This protein interacts with polycystin 1, and they may be partners in a common signaling cascade involved in tubular morphogenesis. Mutations in this gene are associated with autosomal dominant polycystic kidney disease type 2. [provided by RefSeq, Mar 2011]