We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
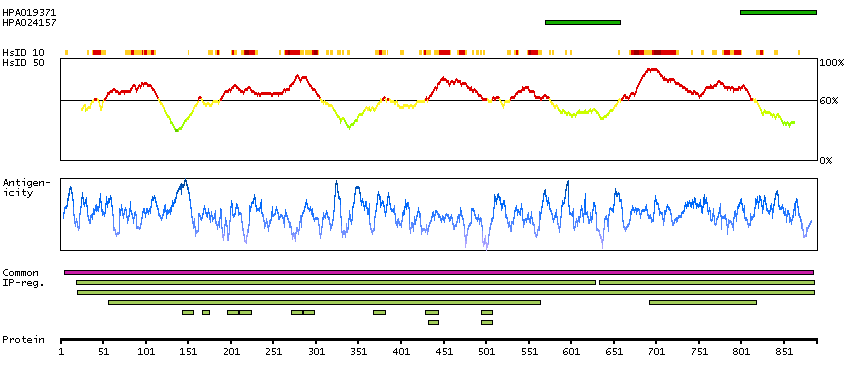
The protein encoded by this gene is a bifunctional, cytosolic protein that functions as an essential enzyme in the TCA cycle and interacts with mRNA to control the levels of iron inside cells. When cellular iron levels are high, this protein binds to a 4Fe-4S cluster and functions as an aconitase. Aconitases are iron-sulfur proteins that function to catalyze the conversion of citrate to isocitrate. When cellular iron levels are low, the protein binds to iron-responsive elements (IREs), which are stem-loop structures found in the 5' UTR of ferritin mRNA, and in the 3' UTR of transferrin receptor mRNA. When the protein binds to IRE, it results in repression of translation of ferritin mRNA, and inhibition of degradation of the otherwise rapidly degraded transferrin receptor mRNA. The encoded protein has been identified as a moonlighting protein based on its ability to perform mechanistically distinct functions. Alternative splicing results in multiple transcript variants [provided by RefSeq, Jan 2014]
Enzymes ENZYME proteins Lyases SPOCTOPUS predicted membrane proteins Predicted intracellular proteins Plasma proteins Citric acid cycle related proteins Cancer-related genes Mutated cancer genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)