We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
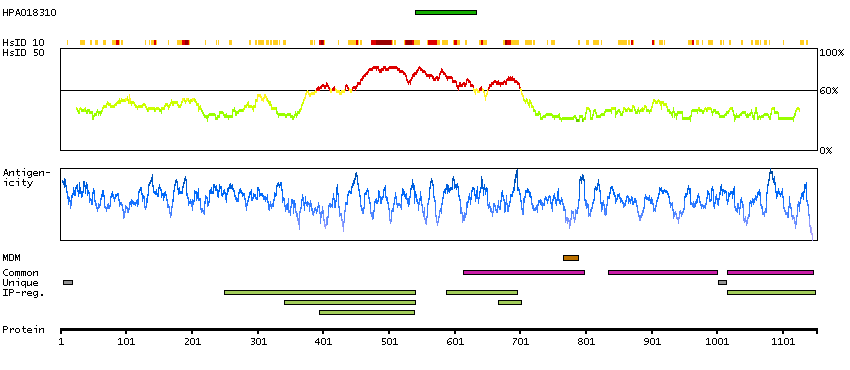
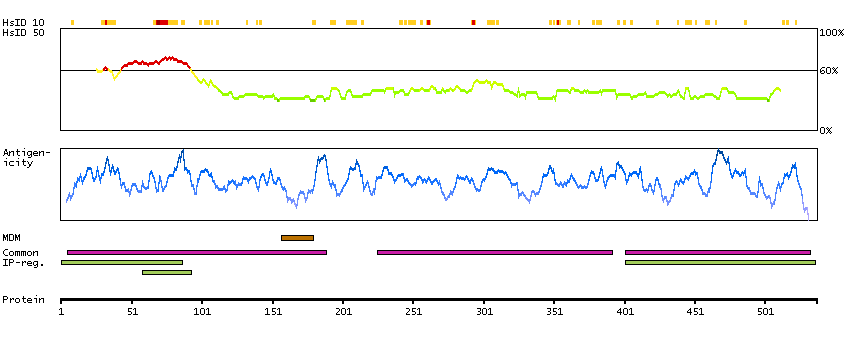
This gene encodes a transcription factor that is required for central nervous system myelination and may regulate oligodendrocyte differentiation. It is thought to act by increasing the expression of genes that effect myelin production but may also directly promote myelin gene expression. Loss of a similar gene in mouse models results in severe demyelination. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Nov 2014]