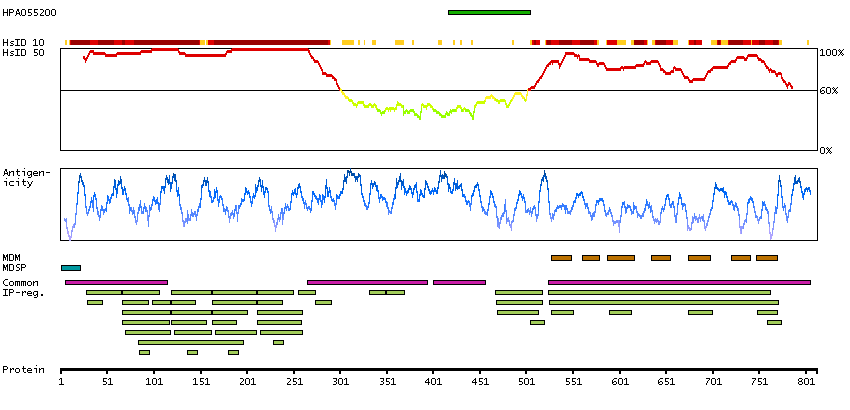
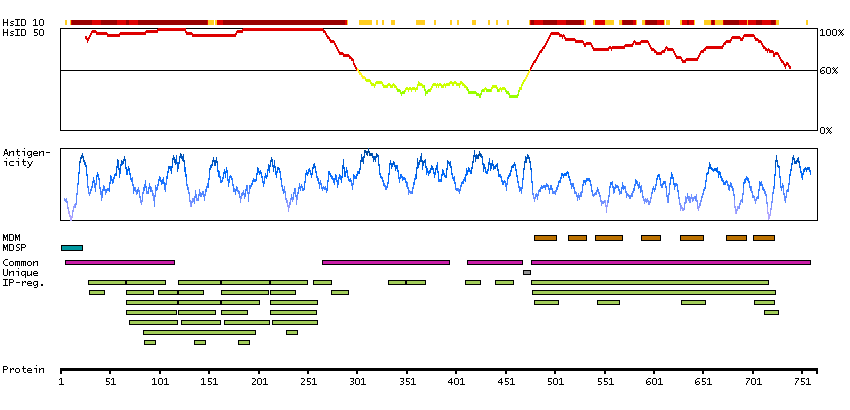
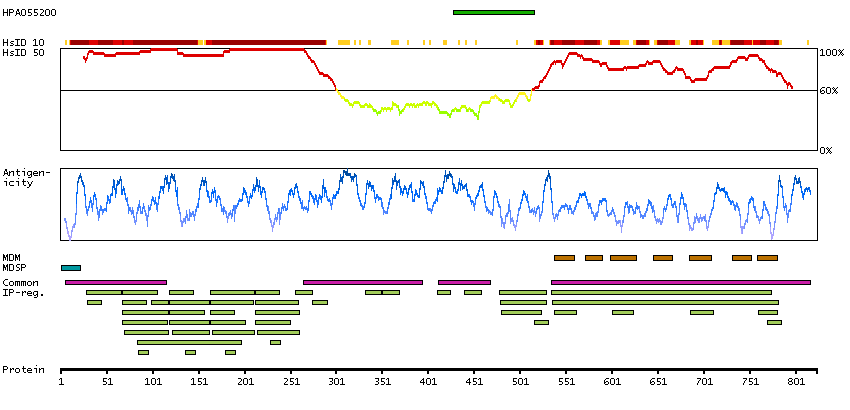
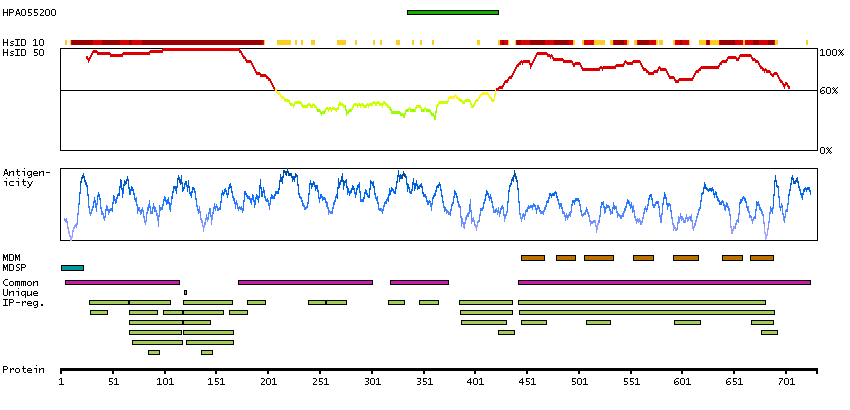
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
RNA tissue category: Group enriched (lung, spleen)
FANTOM5 dataset
Organ
Expression
Alphabetical
RNA tissue category: Expressed in all
GENE INFORMATION
Gene name
ADGRE2 (HGNC Symbol)
Synonyms
CD312, EMR2
Description
Adhesion G protein-coupled receptor E2 (HGNC Symbol)
Entrez gene summary
This gene encodes a member of the class B seven-span transmembrane (TM7) subfamily of G-protein coupled receptors. These proteins are characterized by an extended extracellular region with a variable number of N-terminal epidermal growth factor-like domains coupled to a TM7 domain via a mucin-like spacer domain. The encoded protein is expressed mainly in myeloid cells where it promotes cell-cell adhesion through interaction with chondroitin sulfate chains. This gene is situated in a cluster of related genes on chromosome 19. Alternatively spliced transcript variants encoding multiple isoforms have been observed for this gene. [provided by RefSeq, Aug 2012]