We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
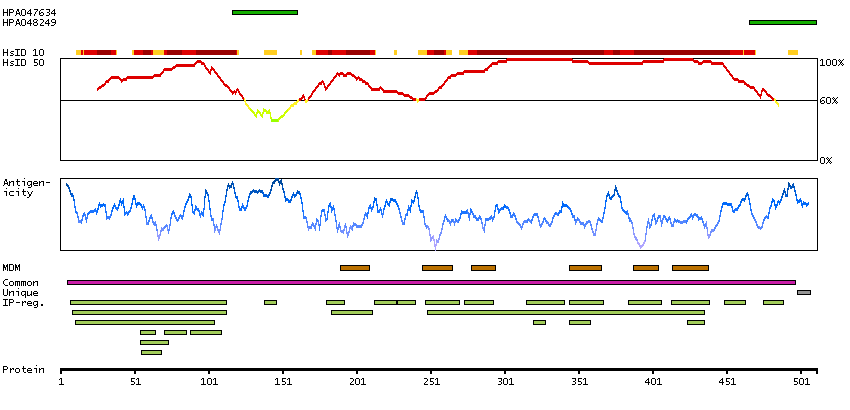
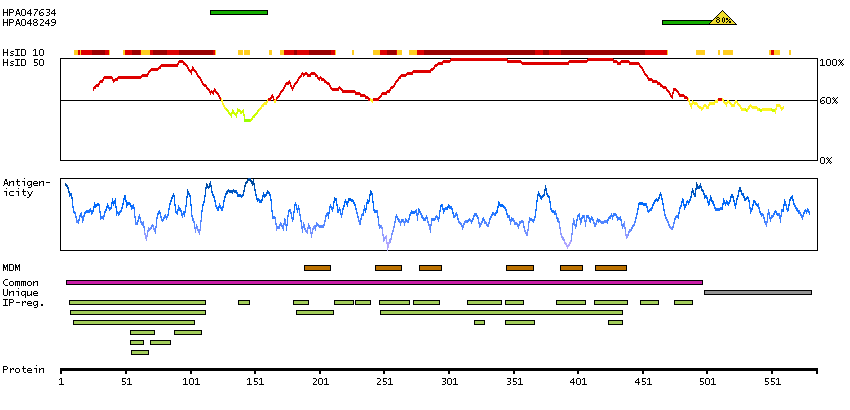
Potassium channel, voltage gated Shaw related subfamily C, member 1 (HGNC Symbol)
Entrez gene summary
This gene encodes a member of a family of integral membrane proteins that mediate the voltage-dependent potassium ion permeability of excitable membranes. Alternative splicing is thought to result in two transcript variants encoding isoforms that differ at their C-termini. These isoforms have had conflicting names in the literature: the longer isoform has been called both ""b"" and ""alpha"", while the shorter isoform has been called both ""a"" and ""beta"" (PMIDs 1432046, 12091563). [provided by RefSeq, Oct 2014]