We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
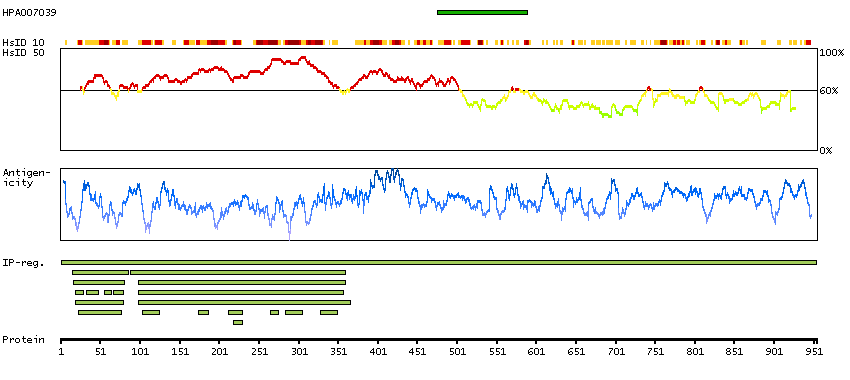
Mitogen-activated protein kinase kinase kinase 10 (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene is a member of the serine/threonine kinase family. This kinase has been shown to activate MAPK8/JNK and MKK4/SEK1, and this kinase itself can be phoshorylated, and thus activated by JNK kinases. This kinase functions preferentially on the JNK signaling pathway, and is reported to be involved in nerve growth factor (NGF) induced neuronal apoptosis. [provided by RefSeq, Jul 2008]