We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
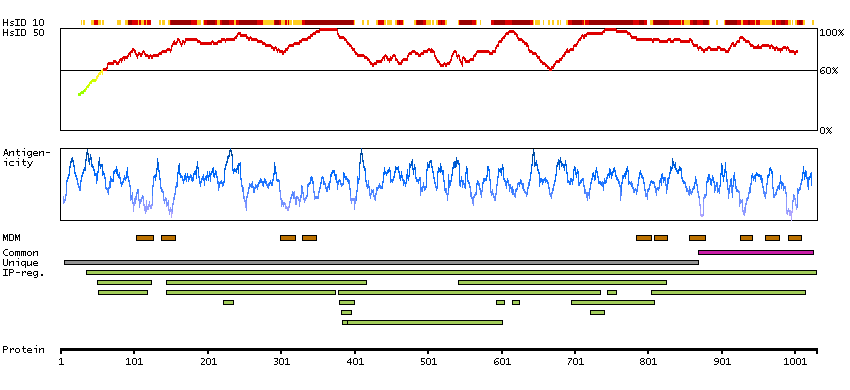
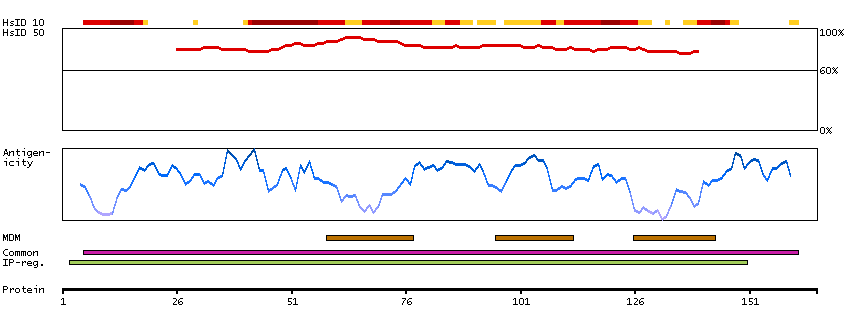
The protein encoded by this gene belongs to the family of P-type cation transport ATPases, and to the subfamily of Na+/K+ -ATPases. Na+/K+ -ATPase is an integral membrane protein responsible for establishing and maintaining the electrochemical gradients of Na and K ions across the plasma membrane. These gradients are essential for osmoregulation, for sodium-coupled transport of a variety of organic and inorganic molecules, and for electrical excitability of nerve and muscle. This enzyme is composed of two subunits, a large catalytic subunit (alpha) and a smaller glycoprotein subunit (beta). The catalytic subunit of Na+/K+ -ATPase is encoded by multiple genes. This gene encodes an alpha 4 subunit. Alternatively spliced transcript variants encoding different isoforms have been identified. [provided by RefSeq, Jul 2008]