We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
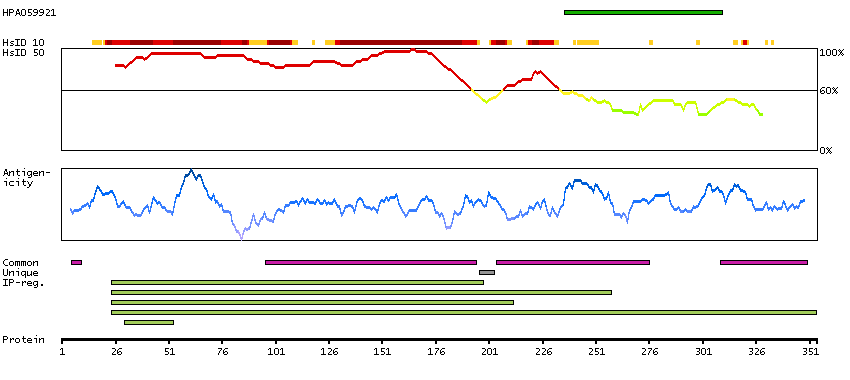
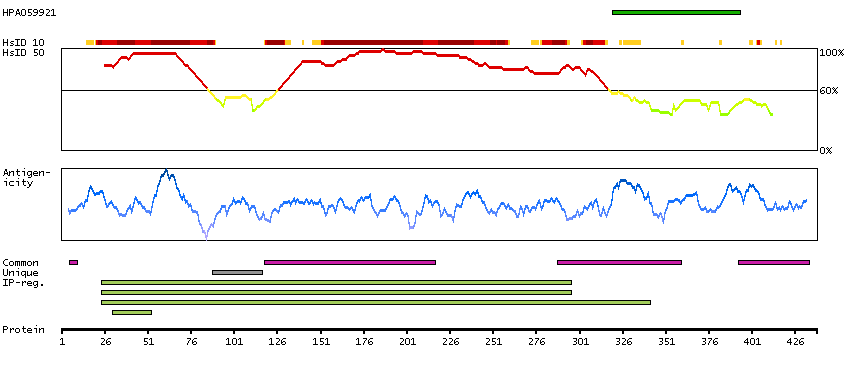
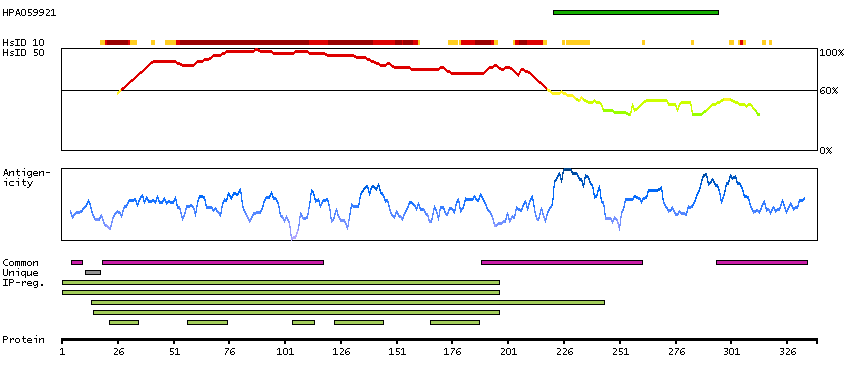
The product of this gene is a member of the GCK group III family of kinases, which are a subset of the Ste20-like kinases. The encoded protein contains an amino-terminal kinase domain, and a carboxy-terminal regulatory domain that mediates homodimerization. The protein kinase localizes to the Golgi apparatus and is specifically activated by binding to the Golgi matrix protein GM130. It is also cleaved by caspase-3 in vitro, and may function in the apoptotic pathway. Several alternatively spliced transcript variants of this gene have been described, but the full-length nature of some of these variants has not been determined. [provided by RefSeq, Jul 2008]
Enzymes ENZYME proteins Transferases Kinases STE Ser/Thr protein kinases Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Enzymes ENZYME proteins Transferases Kinases STE Ser/Thr protein kinases Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Enzymes ENZYME proteins Transferases Kinases STE Ser/Thr protein kinases Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)