We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
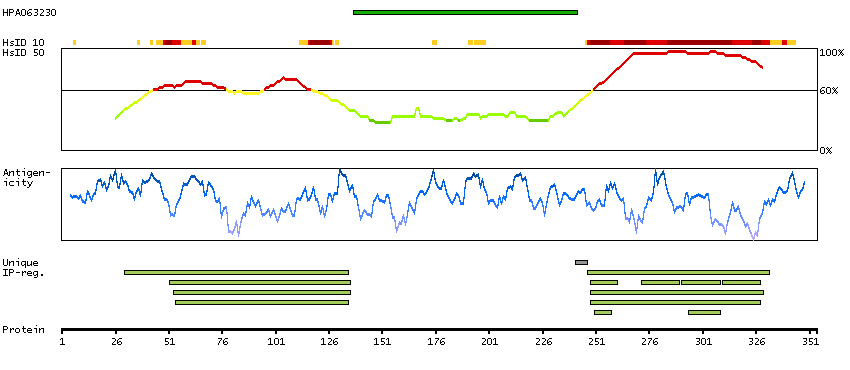
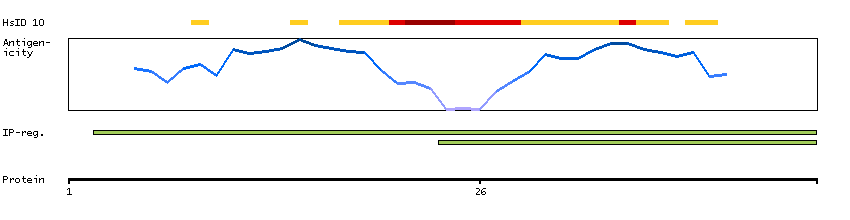
This gene encodes a member of the ETS family of transcription factors, which are defined by the presence of a conserved ETS DNA-binding domain that recognizes the core consensus DNA sequence GGAA/T in target genes. These proteins function either as transcriptional activators or repressors of numerous genes, and are involved in stem cell development, cell senescence and death, and tumorigenesis. Alternatively spliced transcript variants encoding different isoforms have been described for this gene.[provided by RefSeq, Jul 2011]
Predicted intracellular proteins Plasma proteins Transcription factors Helix-turn-helix domains RAS pathway related proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000981 [RNA polymerase II transcription factor activity, sequence-specific DNA binding] GO:0001666 [response to hypoxia] GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005667 [transcription factor complex] GO:0005737 [cytoplasm] GO:0006351 [transcription, DNA-templated] GO:0006355 [regulation of transcription, DNA-templated] GO:0006357 [regulation of transcription from RNA polymerase II promoter] GO:0006366 [transcription from RNA polymerase II promoter] GO:0006955 [immune response] GO:0007565 [female pregnancy] GO:0008134 [transcription factor binding] GO:0008284 [positive regulation of cell proliferation] GO:0008285 [negative regulation of cell proliferation] GO:0009611 [response to wounding] GO:0009612 [response to mechanical stimulus] GO:0010595 [positive regulation of endothelial cell migration] GO:0010715 [regulation of extracellular matrix disassembly] GO:0021854 [hypothalamus development] GO:0021983 [pituitary gland development] GO:0030154 [cell differentiation] GO:0030335 [positive regulation of cell migration] GO:0030578 [PML body organization] GO:0032355 [response to estradiol] GO:0034616 [response to laminar fluid shear stress] GO:0035035 [histone acetyltransferase binding] GO:0042981 [regulation of apoptotic process] GO:0043565 [sequence-specific DNA binding] GO:0044849 [estrous cycle] GO:0045171 [intercellular bridge] GO:0045648 [positive regulation of erythrocyte differentiation] GO:0045765 [regulation of angiogenesis] GO:0045766 [positive regulation of angiogenesis] GO:0045786 [negative regulation of cell cycle] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046677 [response to antibiotic] GO:0048870 [cell motility] GO:0050728 [negative regulation of inflammatory response] GO:0051272 [positive regulation of cellular component movement] GO:0060055 [angiogenesis involved in wound healing] GO:0070301 [cellular response to hydrogen peroxide] GO:0070555 [response to interleukin-1]
Predicted intracellular proteins Plasma proteins Transcription factors Helix-turn-helix domains RAS pathway related proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000981 [RNA polymerase II transcription factor activity, sequence-specific DNA binding] GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005737 [cytoplasm] GO:0006351 [transcription, DNA-templated] GO:0006355 [regulation of transcription, DNA-templated] GO:0006366 [transcription from RNA polymerase II promoter] GO:0006955 [immune response] GO:0008134 [transcription factor binding] GO:0008285 [negative regulation of cell proliferation] GO:0010595 [positive regulation of endothelial cell migration] GO:0030154 [cell differentiation] GO:0030578 [PML body organization] GO:0042981 [regulation of apoptotic process] GO:0043565 [sequence-specific DNA binding] GO:0045171 [intercellular bridge] GO:0045648 [positive regulation of erythrocyte differentiation] GO:0045765 [regulation of angiogenesis] GO:0045786 [negative regulation of cell cycle] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046677 [response to antibiotic] GO:0048870 [cell motility] GO:0051272 [positive regulation of cellular component movement]
Predicted intracellular proteins Plasma proteins Transcription factors Helix-turn-helix domains RAS pathway related proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000981 [RNA polymerase II transcription factor activity, sequence-specific DNA binding] GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005737 [cytoplasm] GO:0006351 [transcription, DNA-templated] GO:0006366 [transcription from RNA polymerase II promoter] GO:0006955 [immune response] GO:0008134 [transcription factor binding] GO:0008285 [negative regulation of cell proliferation] GO:0010595 [positive regulation of endothelial cell migration] GO:0030154 [cell differentiation] GO:0030578 [PML body organization] GO:0042981 [regulation of apoptotic process] GO:0043565 [sequence-specific DNA binding] GO:0045171 [intercellular bridge] GO:0045648 [positive regulation of erythrocyte differentiation] GO:0045765 [regulation of angiogenesis] GO:0045786 [negative regulation of cell cycle] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046677 [response to antibiotic] GO:0048870 [cell motility] GO:0051272 [positive regulation of cellular component movement]
Predicted intracellular proteins Plasma proteins Transcription factors Helix-turn-helix domains RAS pathway related proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000981 [RNA polymerase II transcription factor activity, sequence-specific DNA binding] GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005737 [cytoplasm] GO:0006351 [transcription, DNA-templated] GO:0006355 [regulation of transcription, DNA-templated] GO:0006366 [transcription from RNA polymerase II promoter] GO:0006955 [immune response] GO:0008134 [transcription factor binding] GO:0008285 [negative regulation of cell proliferation] GO:0010595 [positive regulation of endothelial cell migration] GO:0030154 [cell differentiation] GO:0030578 [PML body organization] GO:0042981 [regulation of apoptotic process] GO:0043565 [sequence-specific DNA binding] GO:0045171 [intercellular bridge] GO:0045648 [positive regulation of erythrocyte differentiation] GO:0045765 [regulation of angiogenesis] GO:0045786 [negative regulation of cell cycle] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046677 [response to antibiotic] GO:0048870 [cell motility] GO:0051272 [positive regulation of cellular component movement]
Predicted intracellular proteins RAS pathway related proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000981 [RNA polymerase II transcription factor activity, sequence-specific DNA binding] GO:0005634 [nucleus] GO:0006357 [regulation of transcription from RNA polymerase II promoter] GO:0030154 [cell differentiation] GO:0043565 [sequence-specific DNA binding]
Predicted intracellular proteins Plasma proteins Transcription factors Helix-turn-helix domains RAS pathway related proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000981 [RNA polymerase II transcription factor activity, sequence-specific DNA binding] GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005737 [cytoplasm] GO:0006351 [transcription, DNA-templated] GO:0006355 [regulation of transcription, DNA-templated] GO:0006366 [transcription from RNA polymerase II promoter] GO:0006955 [immune response] GO:0008134 [transcription factor binding] GO:0008285 [negative regulation of cell proliferation] GO:0010595 [positive regulation of endothelial cell migration] GO:0030154 [cell differentiation] GO:0030578 [PML body organization] GO:0042981 [regulation of apoptotic process] GO:0043565 [sequence-specific DNA binding] GO:0045171 [intercellular bridge] GO:0045648 [positive regulation of erythrocyte differentiation] GO:0045765 [regulation of angiogenesis] GO:0045786 [negative regulation of cell cycle] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046677 [response to antibiotic] GO:0048870 [cell motility] GO:0051272 [positive regulation of cellular component movement]