We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
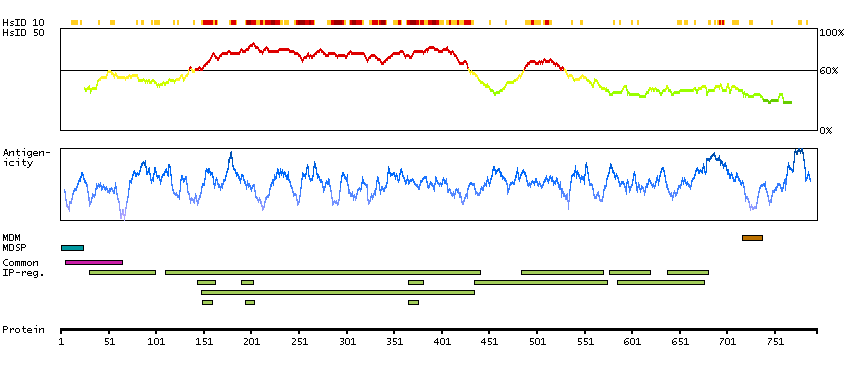
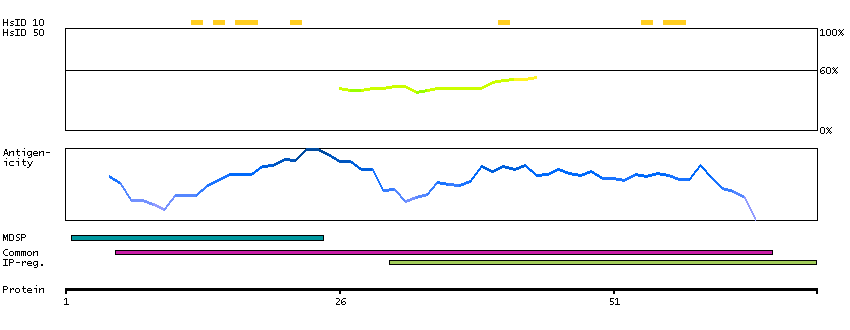
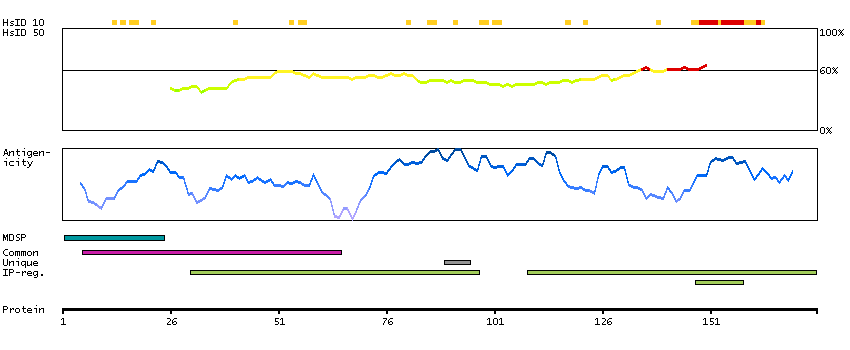
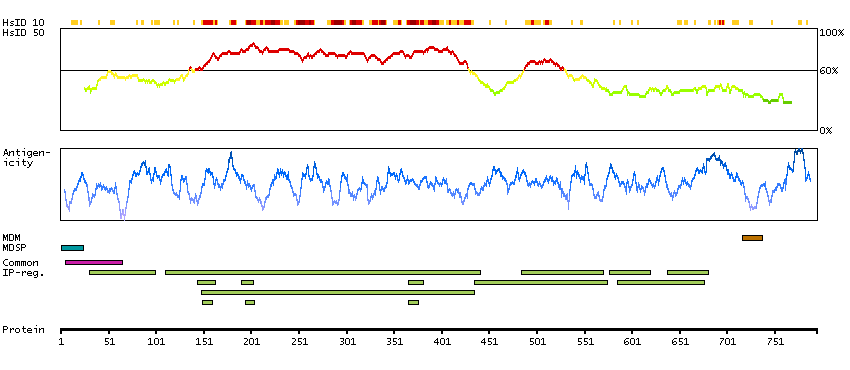
This gene encodes a member of the subtilisin-like proprotein convertase family, which includes proteases that process protein and peptide precursors trafficking through regulated or constitutive branches of the secretory pathway. It encodes a type 1 membrane bound protease that is expressed in many tissues, including neuroendocrine, liver, gut, and brain. The encoded protein undergoes an initial autocatalytic processing event in the ER and then sorts to the trans-Golgi network through endosomes where a second autocatalytic event takes place and the catalytic activity is acquired. The product of this gene is one of the seven basic amino acid-specific members which cleave their substrates at single or paired basic residues. Some of its substrates include proparathyroid hormone, transforming growth factor beta 1 precursor, proalbumin, pro-beta-secretase, membrane type-1 matrix metalloproteinase, beta subunit of pro-nerve growth factor and von Willebrand factor. It is also thought to be one of the proteases responsible for the activation of HIV envelope glycoproteins gp160 and gp140 and may play a role in tumor progression. This gene is located in close proximity to family member proprotein convertase subtilisin/kexin type 6 and upstream of the FES oncogene. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jan 2014]