We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
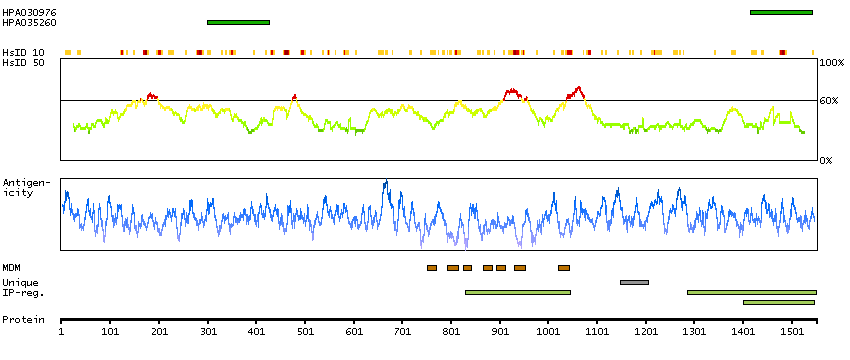
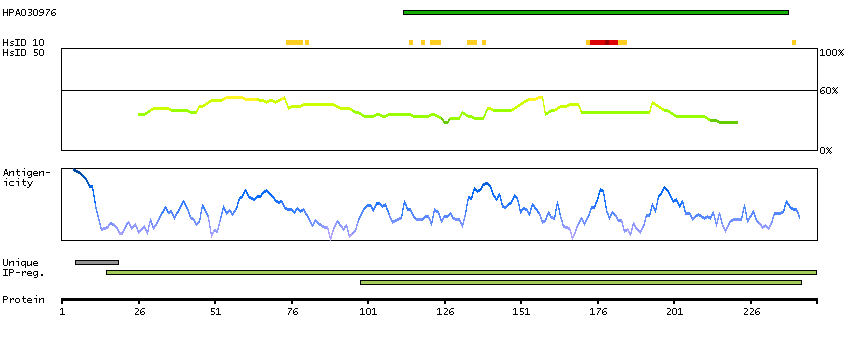
Transient receptor potential cation channel, subfamily M, member 2 (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene is a calcium-permeable cation channel that is regulated by free intracellular ADP-ribose. The encoded protein is activated by oxidative stress and confers susceptibility to cell death. Several alternatively spliced transcript variants of this gene have been described, but their full-length nature is not known. [provided by RefSeq, Jul 2008]