We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
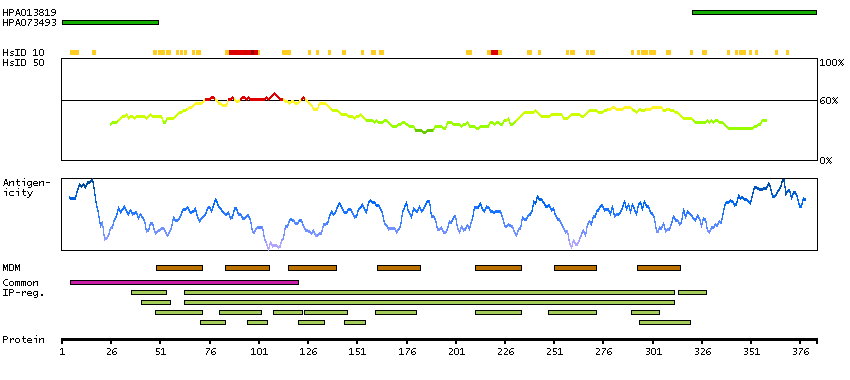
This gene encodes a beta chemokine receptor, which is predicted to be a seven transmembrane protein similar to G protein-coupled receptors. Chemokines and their receptor-mediated signal transduction are critical for the recruitment of effector immune cells to the inflammation site. This gene is expressed in a range of tissues and hemopoietic cells. The expression of this receptor in lymphatic endothelial cells and overexpression in vascular tumors suggested its function in chemokine-driven recirculation of leukocytes and possible chemokine effects on the development and growth of vascular tumors. This receptor appears to bind the majority of beta-chemokine family members; however, its specific function remains unknown. This gene is mapped to chromosome 3p21.3, a region that includes a cluster of chemokine receptor genes. [provided by RefSeq, Jul 2008]