We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Interaction of a cell with the extracellular matrix triggers integrin cell surface receptors to begin signaling cascades that regulate the organization of the actin-cytoskeleton. One of the proteins involved in these cascades is focal adhesion kinase. The protein encoded by this gene is a GTPase activating protein that binds to focal adhesion kinase and mediates the activity of the GTP binding proteins RhoA and Cdc42. Defects in this gene are a cause of juvenile myelomonocytic leukemia (JMML). Two transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Mar 2010]
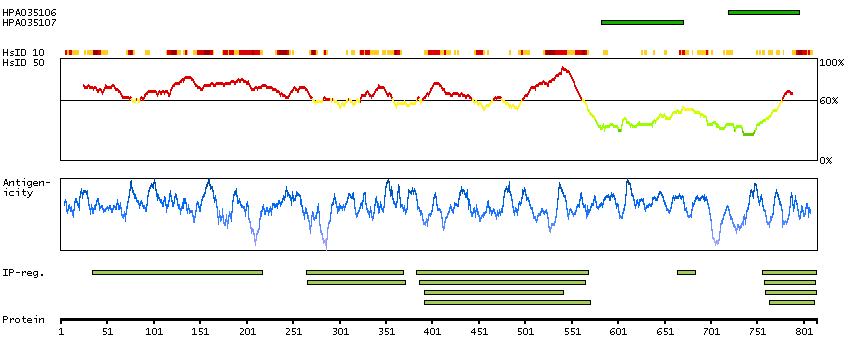
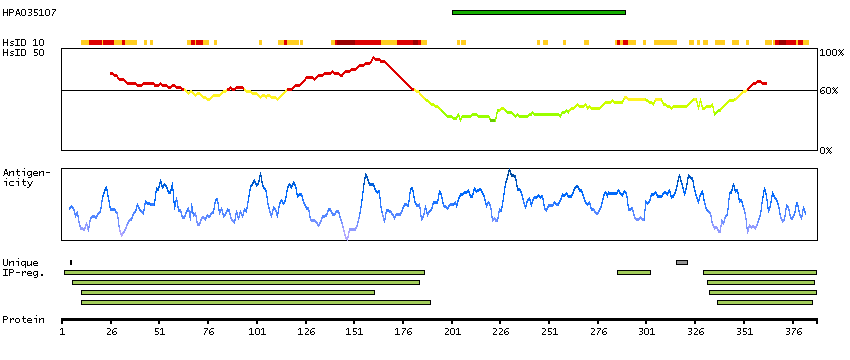
Q9UNA1 [Direct mapping] Rho GTPase-activating protein 26
Show all
Phobius predicted membrane proteins THUMBUP predicted membrane proteins Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Splicing Mutations COSMIC Somatic Mutations COSMIC Frameshift Mutations COSMIC Translocations Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
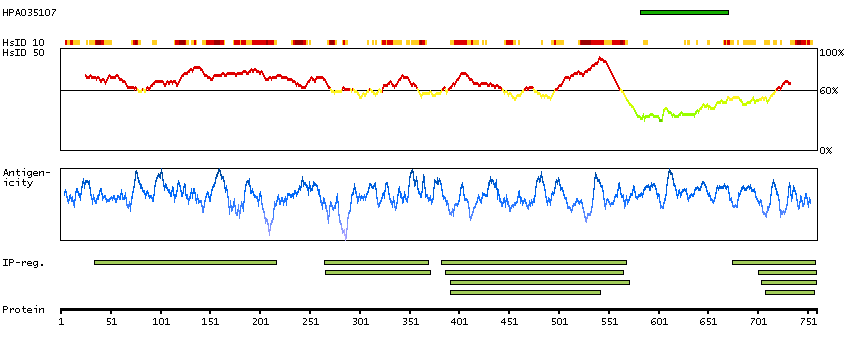
Q9UNA1 [Direct mapping] Rho GTPase-activating protein 26
Show all
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Splicing Mutations COSMIC Somatic Mutations COSMIC Frameshift Mutations COSMIC Translocations Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)