We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
This gene encodes a protein that is one of the components of the Mediator co-activator complex. The Mediator complex is a multi-protein complex required for transcriptional activation by DNA binding transcription factors of genes transcribed by RNA polymerase II. The protein encoded by this gene is similar to cyclin-dependent kinase 8 which can also be a component of the Mediator complex. Alternative splicing results in multiple transcript variants encoding distinct isoforms. [provided by RefSeq, Jul 2014]
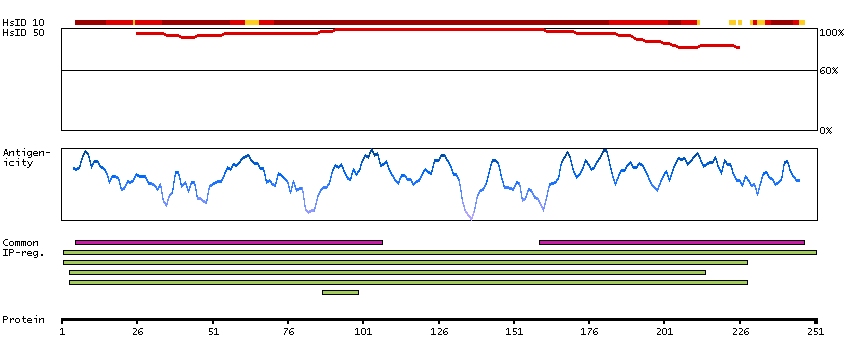
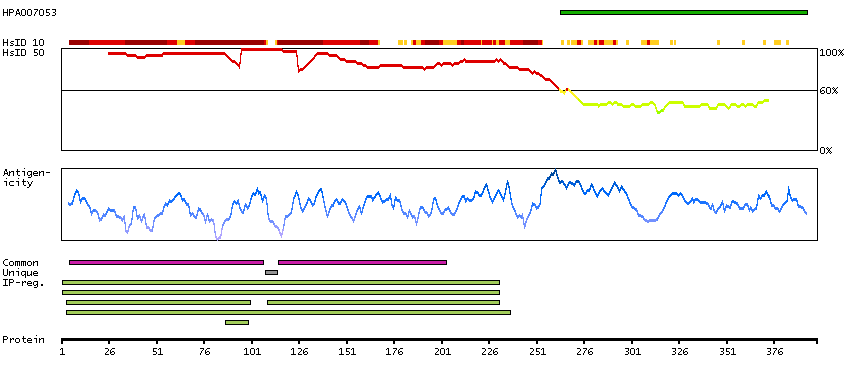
Enzymes ENZYME proteins Transferases Kinases CMGC Ser/Thr protein kinases Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0004672 [protein kinase activity] GO:0004693 [cyclin-dependent protein serine/threonine kinase activity] GO:0005524 [ATP binding] GO:0005634 [nucleus] GO:0005829 [cytosol] GO:0006468 [protein phosphorylation] GO:0019899 [enzyme binding] GO:0043065 [positive regulation of apoptotic process] GO:0050729 [positive regulation of inflammatory response] GO:0051726 [regulation of cell cycle] GO:0071222 [cellular response to lipopolysaccharide]