We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
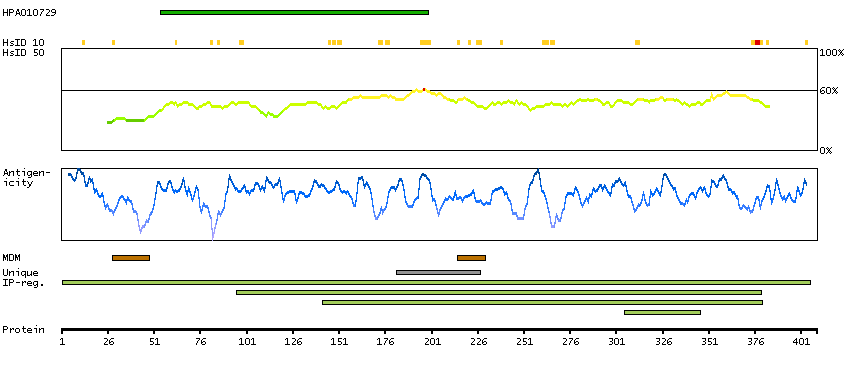
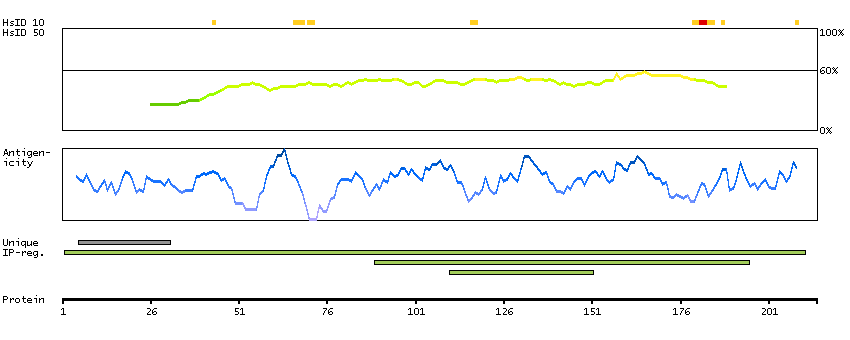
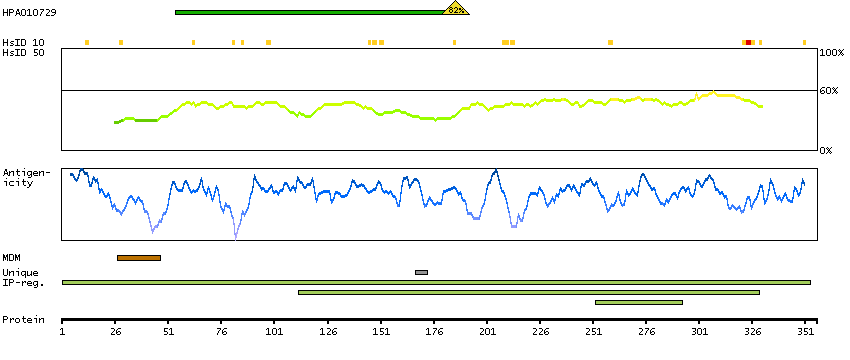
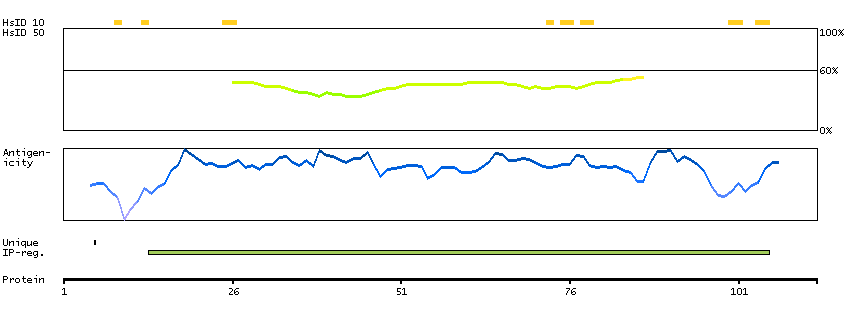
This gene encodes a protein containing an alpha/beta hydrolase fold, which is a catalytic domain found in a very wide range of enzymes. The function of this protein has not been determined. [provided by RefSeq, Jul 2008]