We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
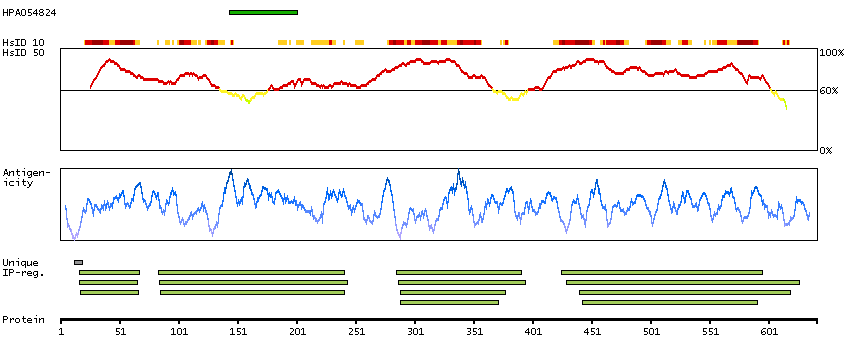
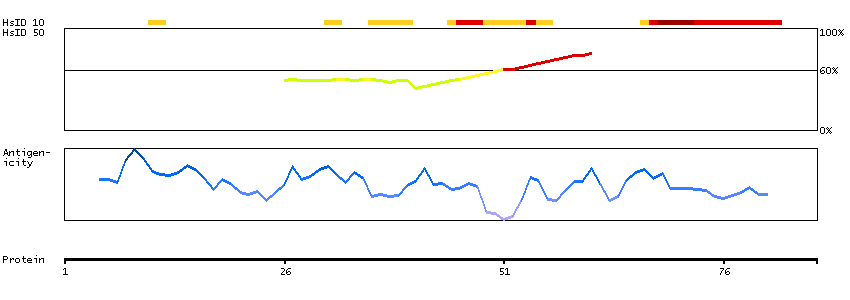
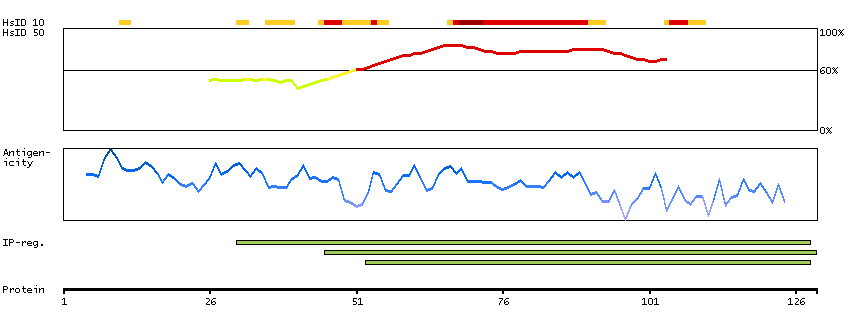
This gene encodes a protein that is similar to the protein encoded by the breakpoint cluster region gene located on chromosome 22. The protein encoded by this gene contains a GTPase-activating protein domain, a domain found in members of the Rho family of GTP-binding proteins. Functional studies in mice determined that this protein plays a role in vestibular morphogenesis. Alternatively spliced transcript variants have been reported for this gene. [provided by RefSeq, Feb 2012]
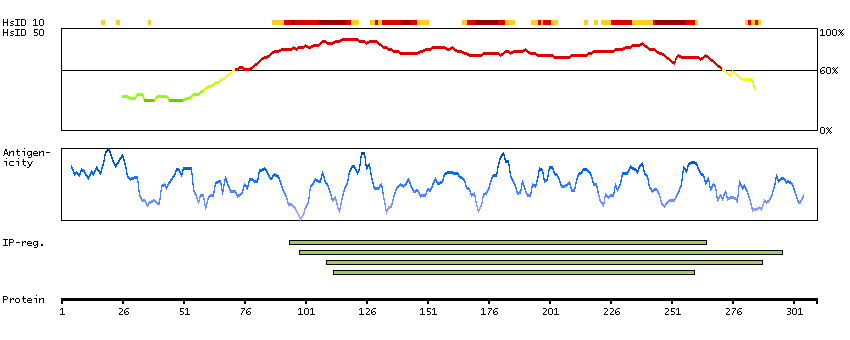
I3L1U8 [Direct mapping] Active breakpoint cluster region-related protein
Show all
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005089 [Rho guanyl-nucleotide exchange factor activity] GO:0035023 [regulation of Rho protein signal transduction] GO:0043547 [positive regulation of GTPase activity]
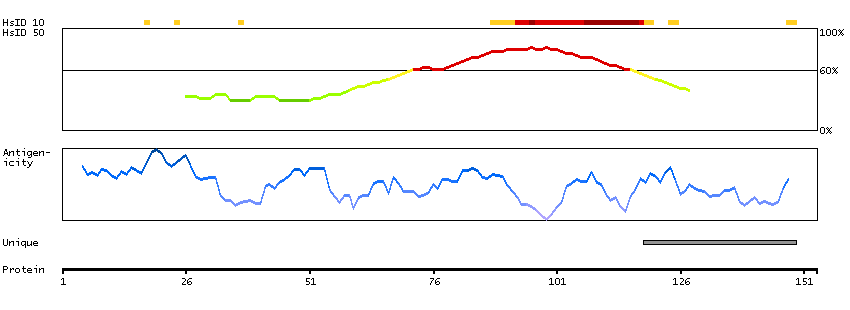
B7Z683 [Direct mapping] Active breakpoint cluster region-related protein; cDNA FLJ54747, highly similar to Active breakpoint cluster region-related protein
Show all
Phobius predicted secreted proteins Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005089 [Rho guanyl-nucleotide exchange factor activity] GO:0005096 [GTPase activator activity] GO:0005515 [protein binding] GO:0007165 [signal transduction] GO:0035023 [regulation of Rho protein signal transduction] GO:0043547 [positive regulation of GTPase activity]
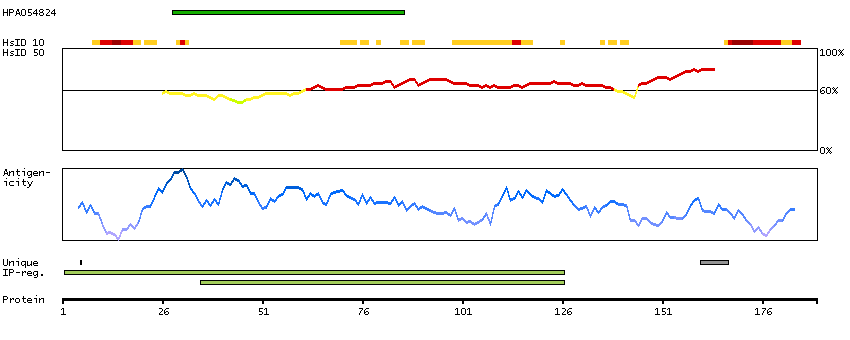
I3L3W0 [Direct mapping] Active breakpoint cluster region-related protein
Show all
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005089 [Rho guanyl-nucleotide exchange factor activity] GO:0035023 [regulation of Rho protein signal transduction] GO:0043547 [positive regulation of GTPase activity]
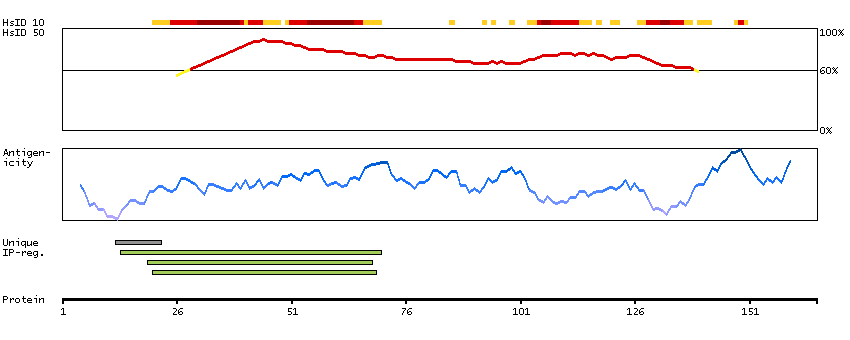
I3L4Y1 [Direct mapping] Active breakpoint cluster region-related protein
Show all
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005089 [Rho guanyl-nucleotide exchange factor activity] GO:0035023 [regulation of Rho protein signal transduction] GO:0043547 [positive regulation of GTPase activity]