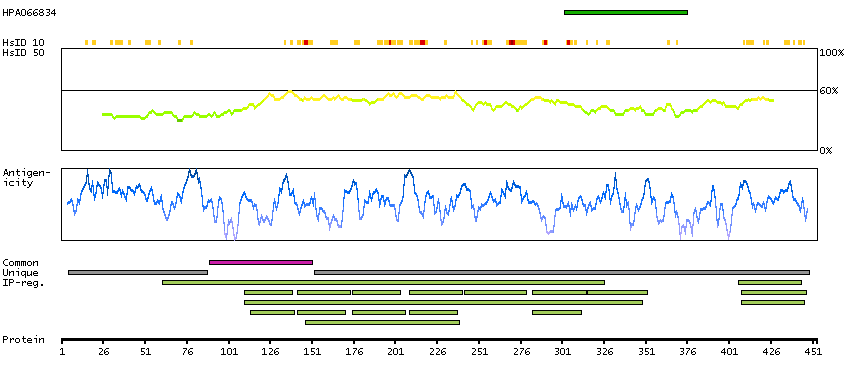
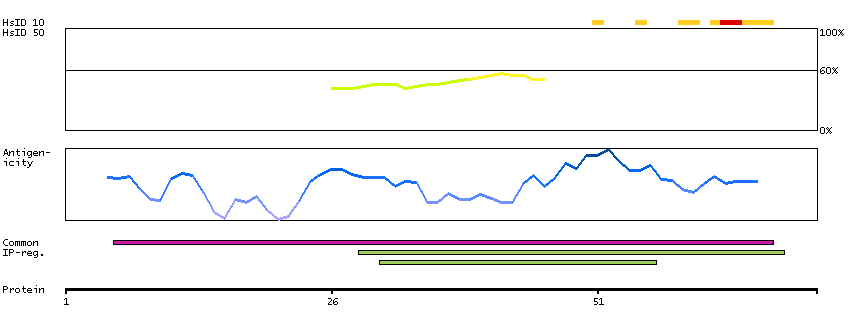
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Ankyrin repeat and SOCS box containing 16 (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene is a member of the ankyrin repeat and SOCS box-containing (ASB) family of proteins. They contain ankyrin repeat sequence and a SOCS box domain. The SOCS box serves to couple suppressor of cytokine signalling (SOCS) proteins and their binding partners with the elongin B and C complex, possibly targeting them for degradation. [provided by RefSeq, Jul 2008]