We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
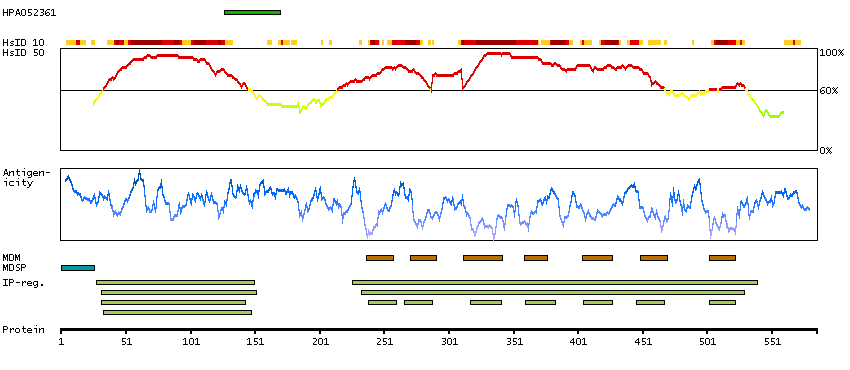
Members of the 'frizzled' gene family encode 7-transmembrane domain proteins that are receptors for Wnt signaling proteins. The FZD5 protein is believed to be the receptor for the Wnt5A ligand. [provided by RefSeq, Jul 2008]