We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
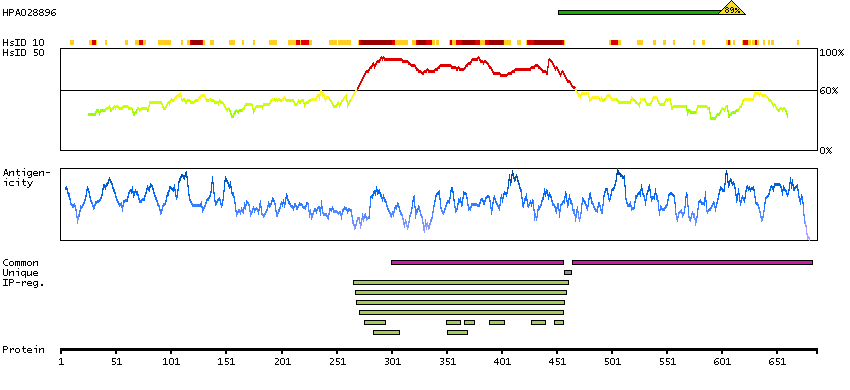
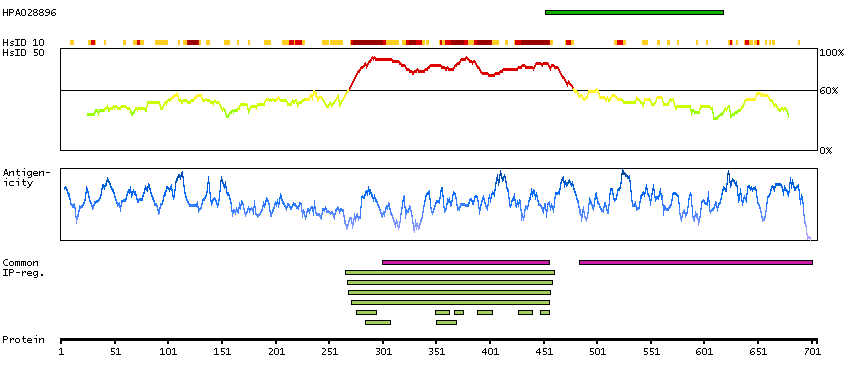
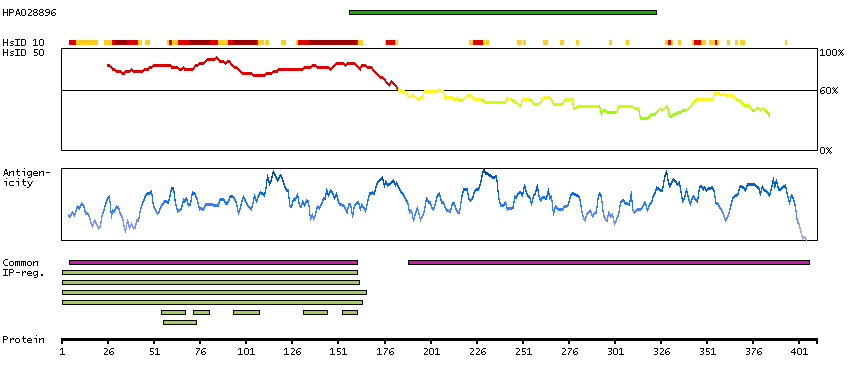
This gene belongs to the TBR1 (T-box brain protein 1) sub-family of T-box genes that share the common DNA-binding T-box domain. The encoded protein is a transcription factor which is crucial for embryonic development of mesoderm and the central nervous system in vertebrates. The protein may also be necessary for the differentiation of effector CD8+ T cells which are involved in defense against viral infections. A similar gene disrupted in mice is shown to be essential during trophoblast development and gastrulation. Alternative splicing results in multiple transcript variants. [provided by RefSeq, May 2013]
Predicted intracellular proteins Transcription factors Immunoglobulin fold Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0001102 [RNA polymerase II activating transcription factor binding] GO:0001191 [transcriptional repressor activity, RNA polymerase II transcription factor binding] GO:0001706 [endoderm formation] GO:0001707 [mesoderm formation] GO:0002250 [adaptive immune response] GO:0002302 [CD8-positive, alpha-beta T cell differentiation involved in immune response] GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005634 [nucleus] GO:0006351 [transcription, DNA-templated] GO:0006355 [regulation of transcription, DNA-templated] GO:0007420 [brain development] GO:0043565 [sequence-specific DNA binding] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0060706 [cell differentiation involved in embryonic placenta development] GO:0060809 [mesodermal to mesenchymal transition involved in gastrulation]