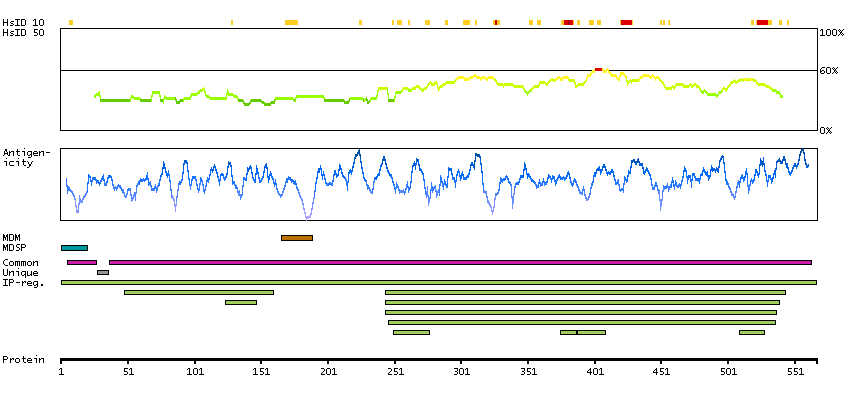
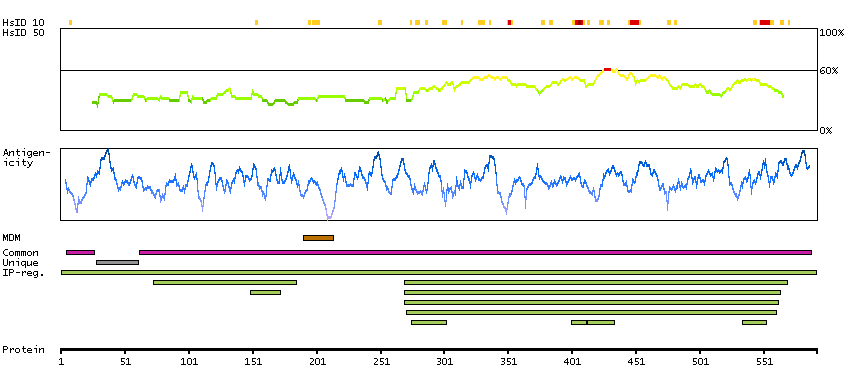
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Transforming growth factor, beta receptor II (70/80kDa) (HGNC Symbol)
Entrez gene summary
This gene encodes a member of the Ser/Thr protein kinase family and the TGFB receptor subfamily. The encoded protein is a transmembrane protein that has a protein kinase domain, forms a heterodimeric complex with another receptor protein, and binds TGF-beta. This receptor/ligand complex phosphorylates proteins, which then enter the nucleus and regulate the transcription of a subset of genes related to cell proliferation. Mutations in this gene have been associated with Marfan Syndrome, Loeys-Deitz Aortic Aneurysm Syndrome, and the development of various types of tumors. Alternatively spliced transcript variants encoding different isoforms have been characterized. [provided by RefSeq, Jul 2008]