We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
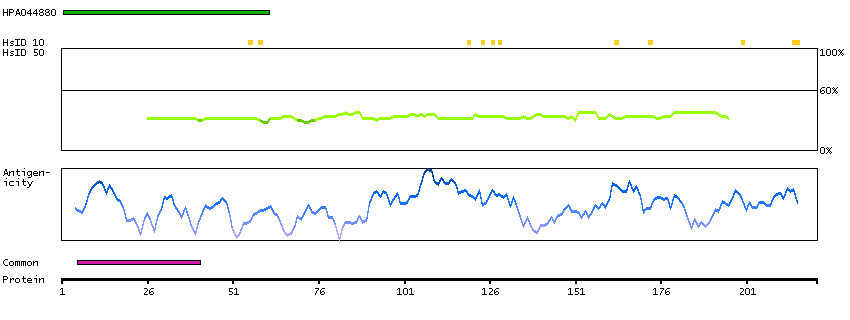
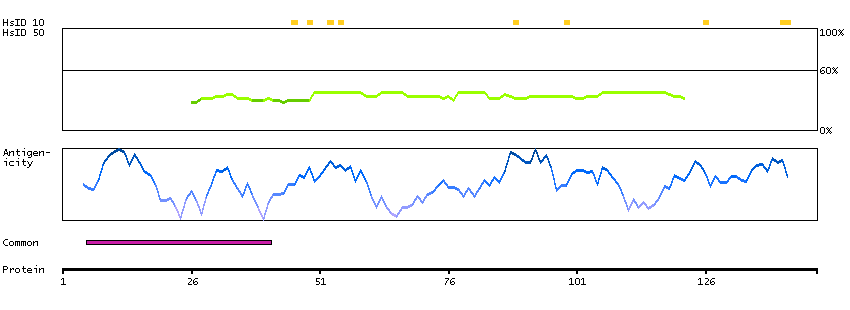
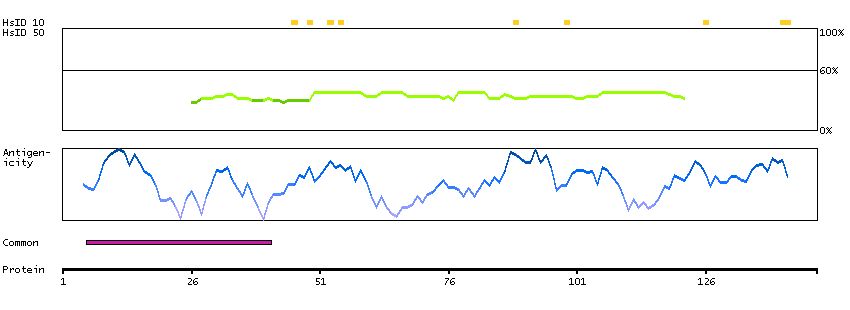
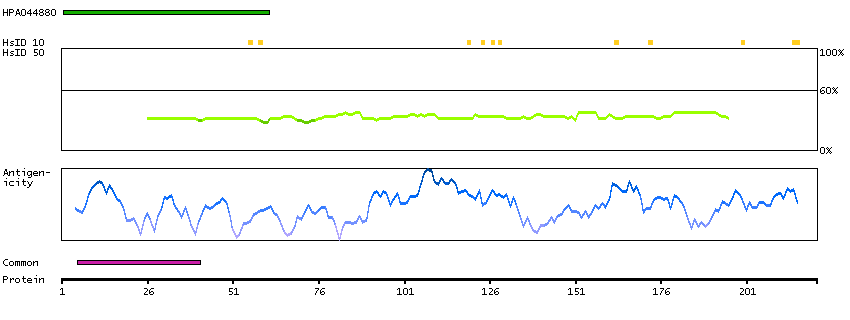
This gene encodes a protein that was identified as a cellular interacting partner of non-structural protein 10 of the severe acute respiratory syndrome coronavirus (SARS-CoV). The encoded protein may function as a negative regulator of transcription. There is a pseudogene for this gene on chromosome 1. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Mar 2013]