We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
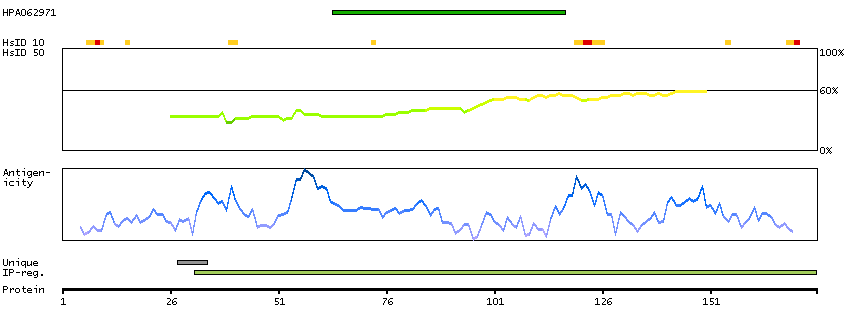
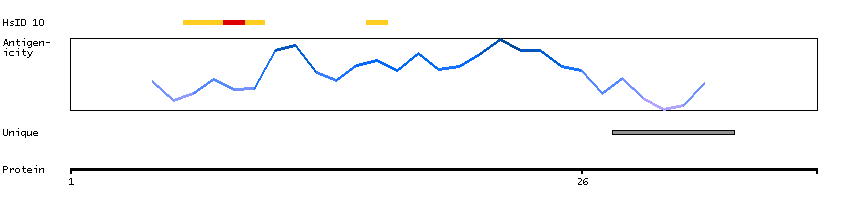
Isocitrate dehydrogenases catalyze the oxidative decarboxylation of isocitrate to 2-oxoglutarate. These enzymes belong to two distinct subclasses, one of which utilizes NAD(+) as the electron acceptor and the other NADP(+). Five isocitrate dehydrogenases have been reported: three NAD(+)-dependent isocitrate dehydrogenases, which localize to the mitochondrial matrix, and two NADP(+)-dependent isocitrate dehydrogenases, one of which is mitochondrial and the other predominantly cytosolic. NAD(+)-dependent isocitrate dehydrogenases catalyze the allosterically regulated rate-limiting step of the tricarboxylic acid cycle. Each isozyme is a heterotetramer that is composed of two alpha subunits, one beta subunit, and one gamma subunit. The protein encoded by this gene is the alpha subunit of one isozyme of NAD(+)-dependent isocitrate dehydrogenase. [provided by RefSeq, Jul 2008]
Enzymes ENZYME proteins Oxidoreductases Predicted intracellular proteins Plasma proteins Citric acid cycle related proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000287 [magnesium ion binding] GO:0004449 [isocitrate dehydrogenase (NAD+) activity] GO:0005634 [nucleus] GO:0005739 [mitochondrion] GO:0005759 [mitochondrial matrix] GO:0005975 [carbohydrate metabolic process] GO:0006099 [tricarboxylic acid cycle] GO:0016616 [oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor] GO:0043209 [myelin sheath] GO:0044237 [cellular metabolic process] GO:0044281 [small molecule metabolic process] GO:0051287 [NAD binding] GO:0055114 [oxidation-reduction process]
Predicted intracellular proteins Citric acid cycle related proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000287 [magnesium ion binding] GO:0005739 [mitochondrion] GO:0016616 [oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor] GO:0051287 [NAD binding] GO:0055114 [oxidation-reduction process]
Predicted intracellular proteins Citric acid cycle related proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000287 [magnesium ion binding] GO:0005739 [mitochondrion] GO:0016616 [oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor] GO:0051287 [NAD binding] GO:0055114 [oxidation-reduction process]
Predicted intracellular proteins Citric acid cycle related proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000287 [magnesium ion binding] GO:0005739 [mitochondrion] GO:0016616 [oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor] GO:0051287 [NAD binding] GO:0055114 [oxidation-reduction process]
Predicted intracellular proteins Citric acid cycle related proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000287 [magnesium ion binding] GO:0016616 [oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor] GO:0051287 [NAD binding] GO:0055114 [oxidation-reduction process]
Predicted intracellular proteins Citric acid cycle related proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000287 [magnesium ion binding] GO:0005739 [mitochondrion] GO:0016616 [oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor] GO:0051287 [NAD binding] GO:0055114 [oxidation-reduction process]
Predicted intracellular proteins Citric acid cycle related proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000287 [magnesium ion binding] GO:0005739 [mitochondrion] GO:0016616 [oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor] GO:0051287 [NAD binding] GO:0055114 [oxidation-reduction process]