We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
This gene was identified based on its homology to the gene encoding the RING3 protein, a serine/threonine kinase. The gene localizes to 9q34, a region which contains several major histocompatibility complex (MHC) genes. The function of the encoded protein is not known. [provided by RefSeq, Jul 2008]
SPOCTOPUS predicted secreted proteins Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
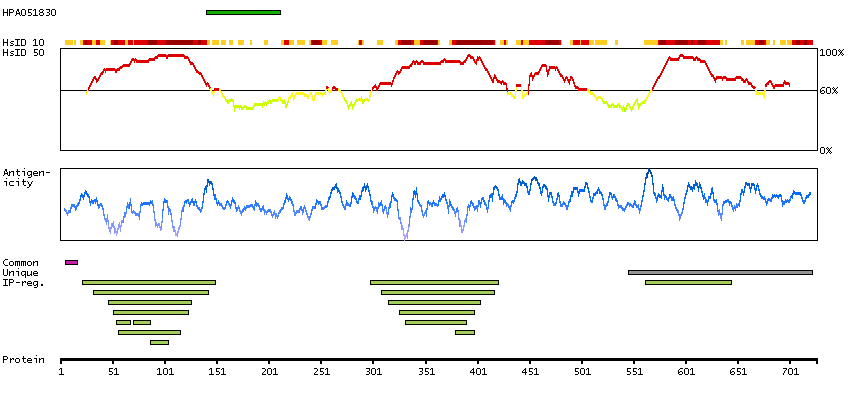
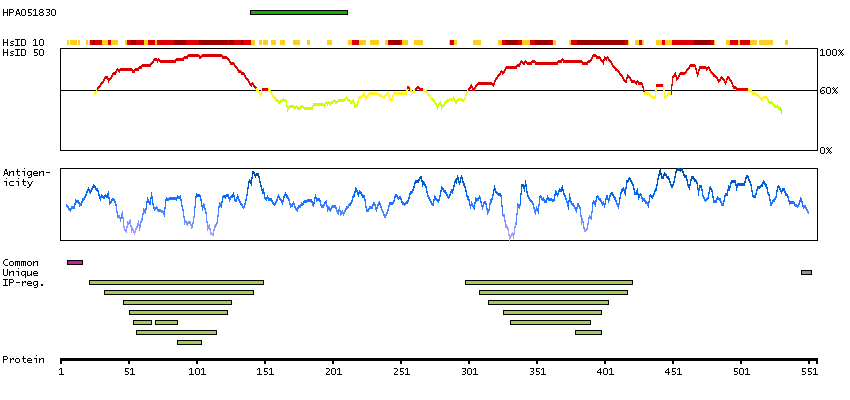
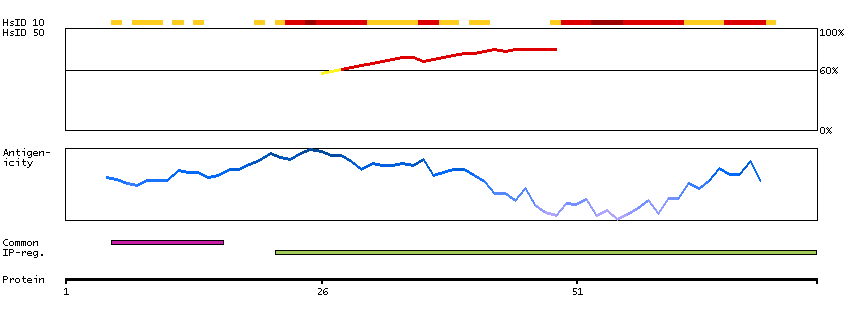
Q15059 [Direct mapping] Bromodomain-containing protein 3
Show all
SPOCTOPUS predicted secreted proteins Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Disease related genes Protein evidence (Ezkurdia et al 2014)