We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Aminoacyl-tRNA synthetases are a class of enzymes that charge tRNAs with their cognate amino acids. The protein encoded by this gene is a cytoplasmic enzyme which belongs to the class II family of aminoacyl-tRNA synthetases. The enzyme is responsible for the synthesis of histidyl-transfer RNA, which is essential for the incorporation of histidine into proteins. The gene is located in a head-to-head orientation with HARSL on chromosome five, where the homologous genes share a bidirectional promoter. The gene product is a frequent target of autoantibodies in the human autoimmune disease polymyositis/dermatomyositis. Several transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Apr 2012]
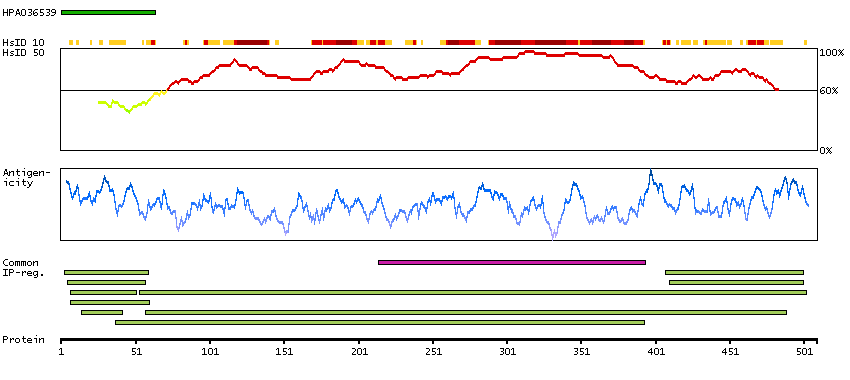
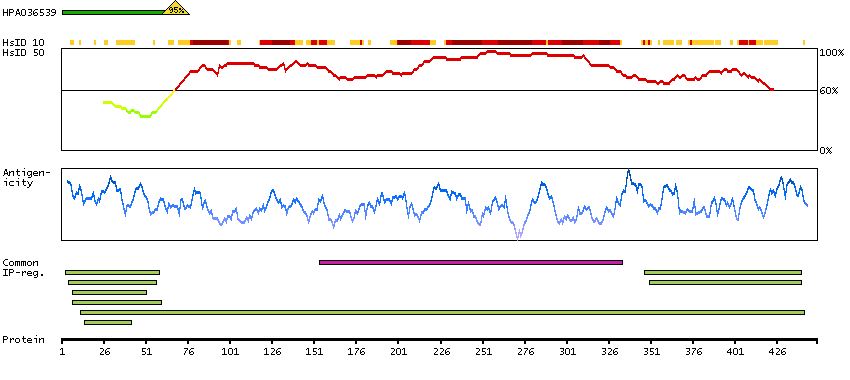
Enzymes ENZYME proteins Ligase Predicted intracellular proteins Plasma proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
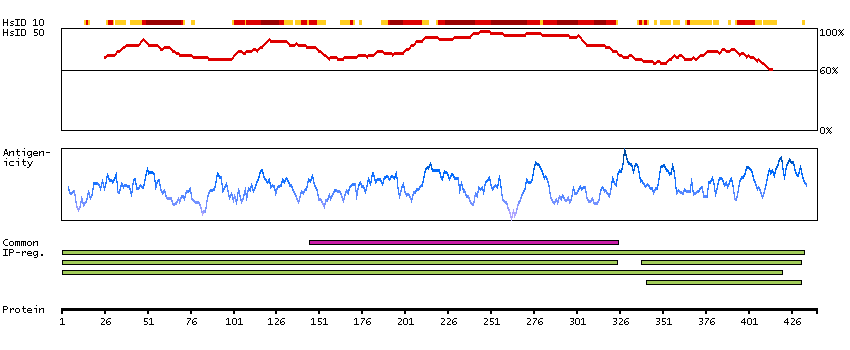
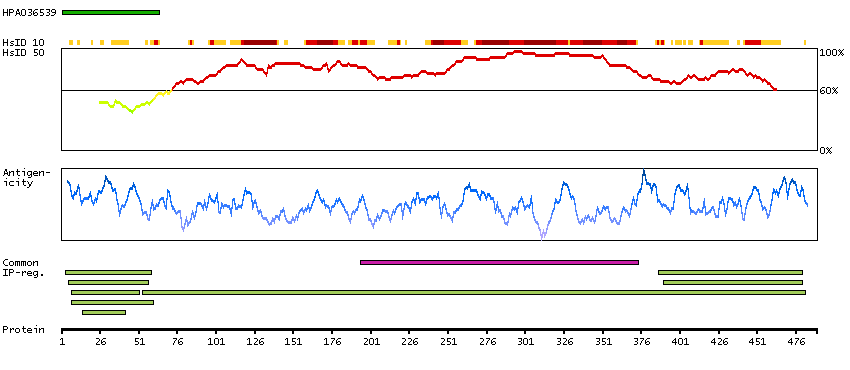
Enzymes ENZYME proteins Ligase Predicted intracellular proteins Plasma proteins Disease related genes Potential drug targets Protein evidence (Ezkurdia et al 2014)
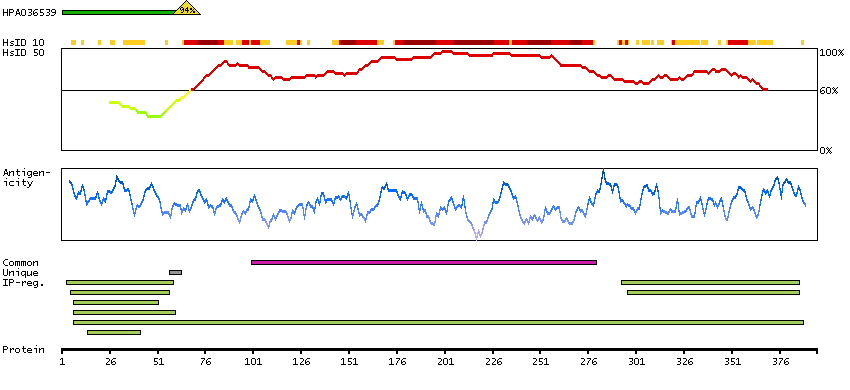
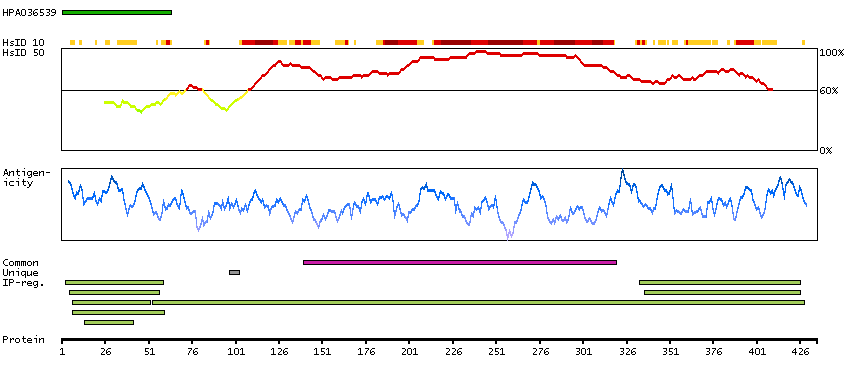
Enzymes ENZYME proteins Ligase Predicted intracellular proteins Plasma proteins Disease related genes Potential drug targets Protein evidence (Ezkurdia et al 2014)
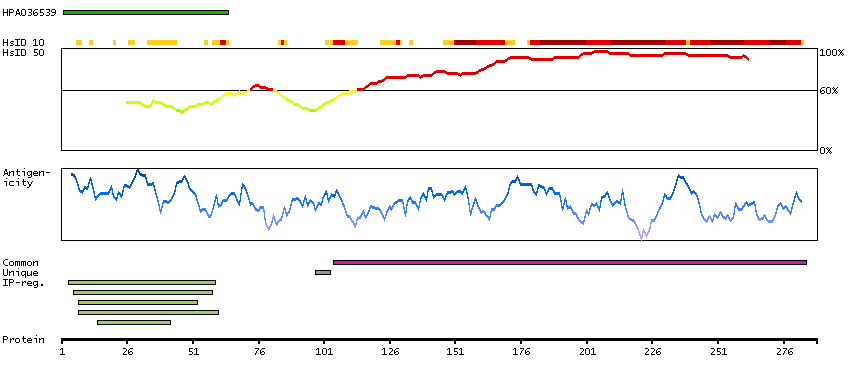
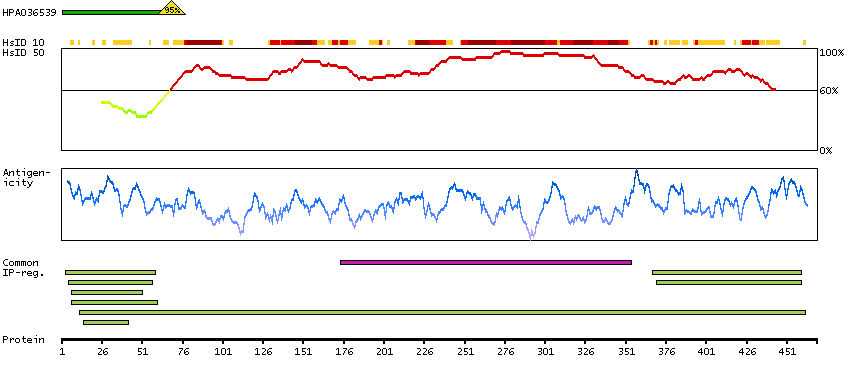
Enzymes ENZYME proteins Ligase Predicted intracellular proteins Plasma proteins Disease related genes Potential drug targets Protein evidence (Ezkurdia et al 2014)