We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Cancer-related genes CD markers Disease related genes Enzymes FDA approved drug targets Plasma proteins Predicted intracellular proteins Predicted membrane proteins
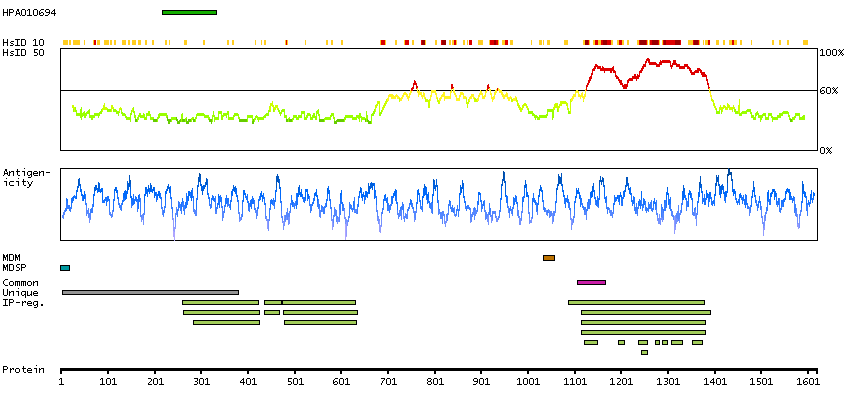
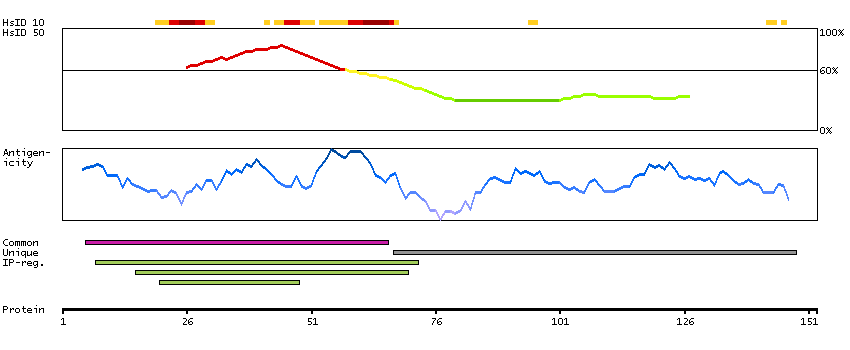
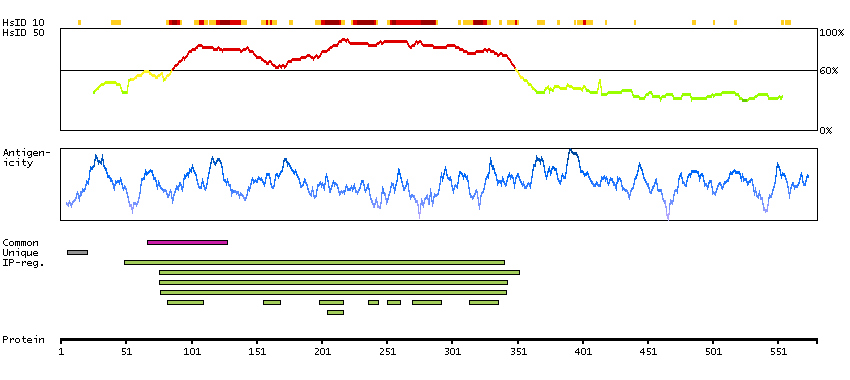
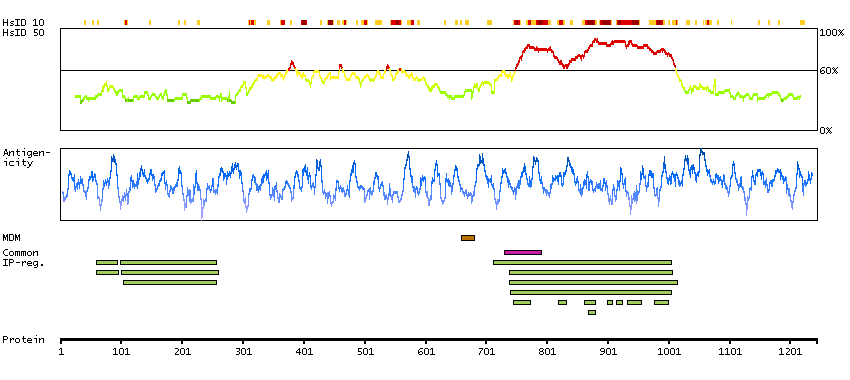
This gene encodes a receptor tyrosine kinase, which belongs to the insulin receptor superfamily. This protein comprises an extracellular domain, an hydrophobic stretch corresponding to a single pass transmembrane region, and an intracellular kinase domain. It plays an important role in the development of the brain and exerts its effects on specific neurons in the nervous system. This gene has been found to be rearranged, mutated, or amplified in a series of tumours including anaplastic large cell lymphomas, neuroblastoma, and non-small cell lung cancer. The chromosomal rearrangements are the most common genetic alterations in this gene, which result in creation of multiple fusion genes in tumourigenesis, including ALK (chromosome 2)/EML4 (chromosome 2), ALK/RANBP2 (chromosome 2), ALK/ATIC (chromosome 2), ALK/TFG (chromosome 3), ALK/NPM1 (chromosome 5), ALK/SQSTM1 (chromosome 5), ALK/KIF5B (chromosome 10), ALK/CLTC (chromosome 17), ALK/TPM4 (chromosome 19), and ALK/MSN (chromosome X).[provided by RefSeq, Jan 2011]