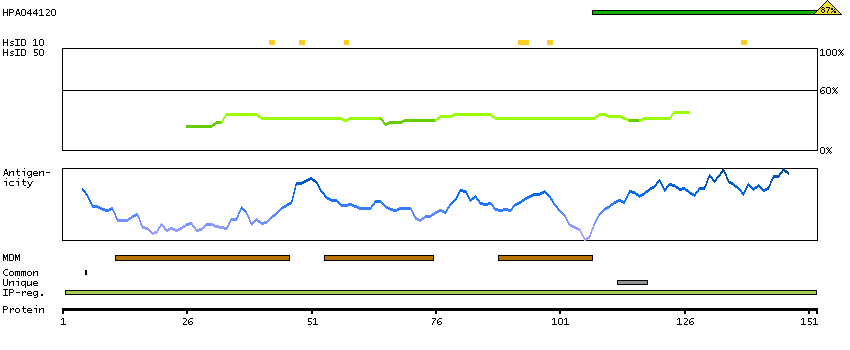
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Angiotensin II receptor-associated protein (HGNC Symbol)
Entrez gene summary
This gene encodes a transmembrane protein localized to the plasma membrane and perinuclear vesicular structures. The gene product interacts with the angiotensin II type I receptor and negatively regulates angiotensin II signaling. Alternative splicing of this gene generates multiple transcript variants encoding different isoforms. [provided by RefSeq, Jul 2008]