We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
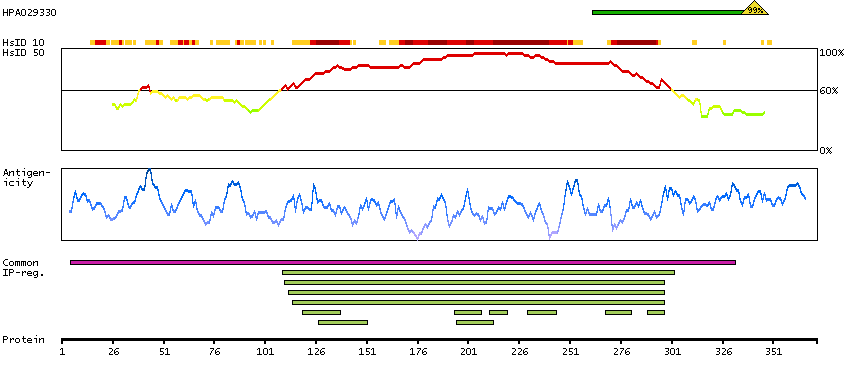
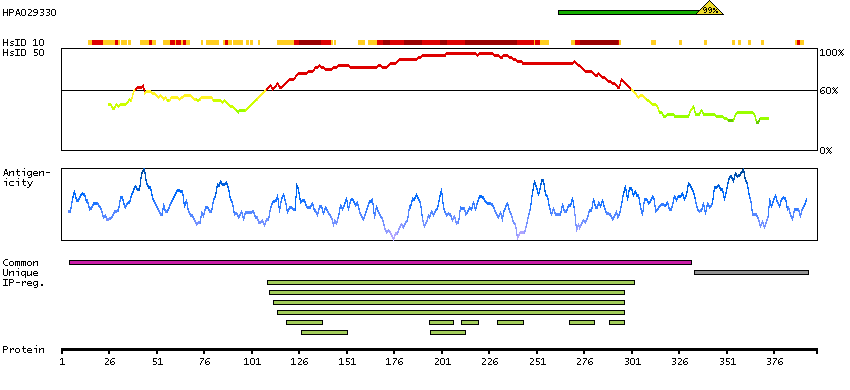
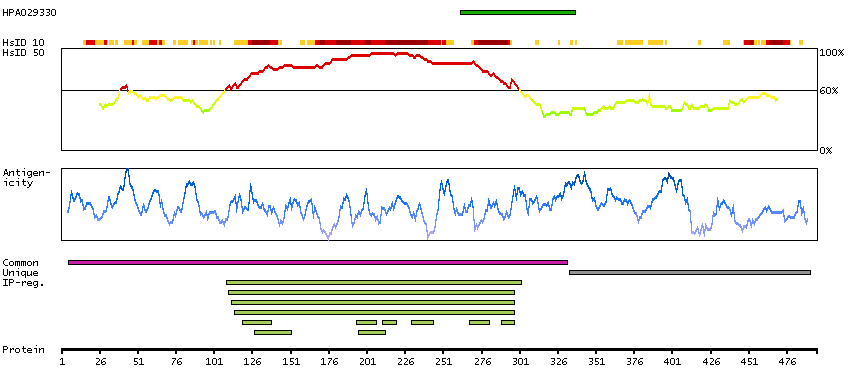
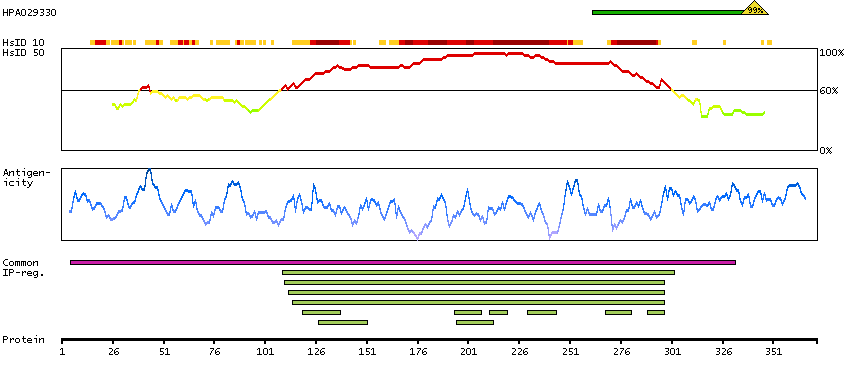
This gene is a member of a phylogenetically conserved family of genes that share a common DNA-binding domain, the T-box. T-box genes encode transcription factors involved in the regulation of developmental processes. This gene product shares 98% amino acid sequence identity with the mouse ortholog. DiGeorge syndrome (DGS)/velocardiofacial syndrome (VCFS), a common congenital disorder characterized by neural-crest-related developmental defects, has been associated with deletions of chromosome 22q11.2, where this gene has been mapped. Studies using mouse models of DiGeorge syndrome suggest a major role for this gene in the molecular etiology of DGS/VCFS. Several alternatively spliced transcript variants encoding different isoforms have been described for this gene. [provided by RefSeq, Jul 2008]