We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
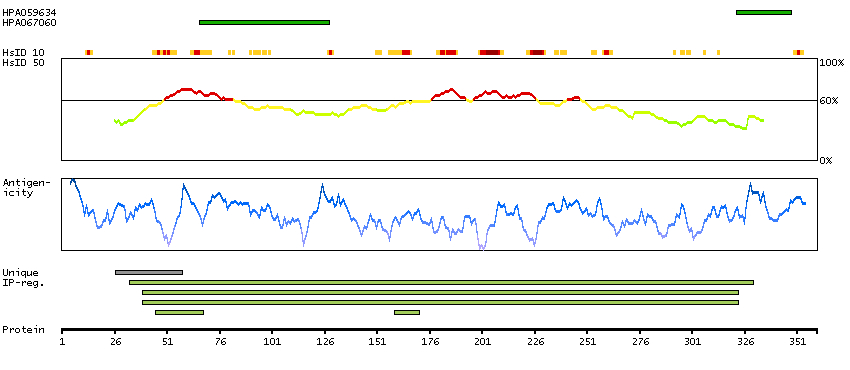
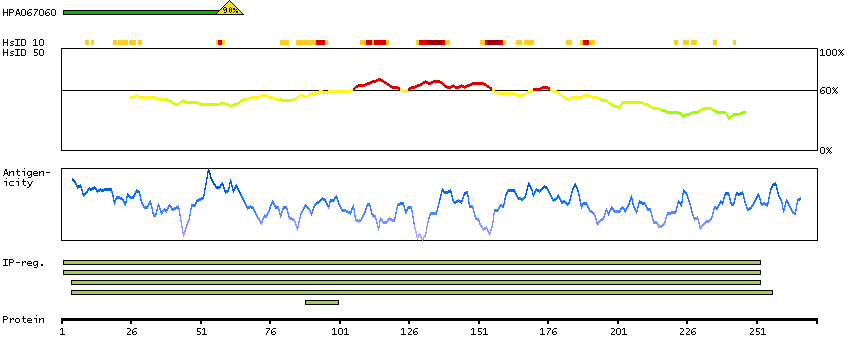
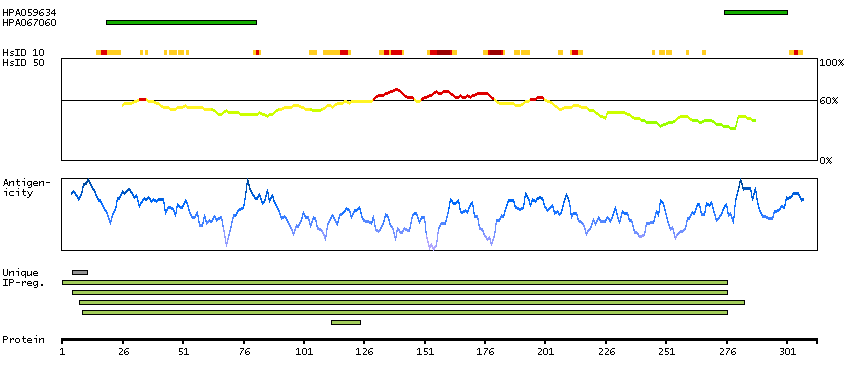
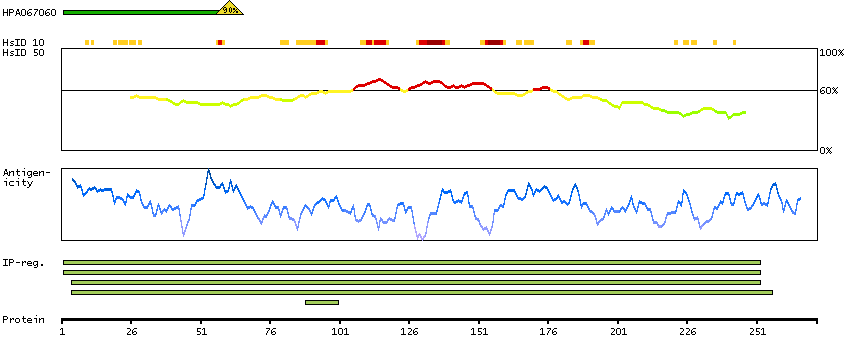
The protein encoded by this gene belongs to the CDK subfamily of the Ser/Thr protein kinase family. The CDK subfamily members are highly similar to the gene products of S. cerevisiae cdc28, and S. pombe cdc2, and are known to be essential for cell cycle progression. This kinase has been shown to play a role in cellular proliferation and its function is limited to cell cycle G2-M phase. Multiple transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, May 2009]
Enzymes ENZYME proteins Transferases Kinases CMGC Ser/Thr protein kinases MEMSAT-SVM predicted membrane proteins Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0004672 [protein kinase activity] GO:0004693 [cyclin-dependent protein serine/threonine kinase activity] GO:0005515 [protein binding] GO:0005524 [ATP binding] GO:0006468 [protein phosphorylation] GO:0007089 [traversing start control point of mitotic cell cycle] GO:0008285 [negative regulation of cell proliferation] GO:0043410 [positive regulation of MAPK cascade] GO:0097472 [cyclin-dependent protein kinase activity]