We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
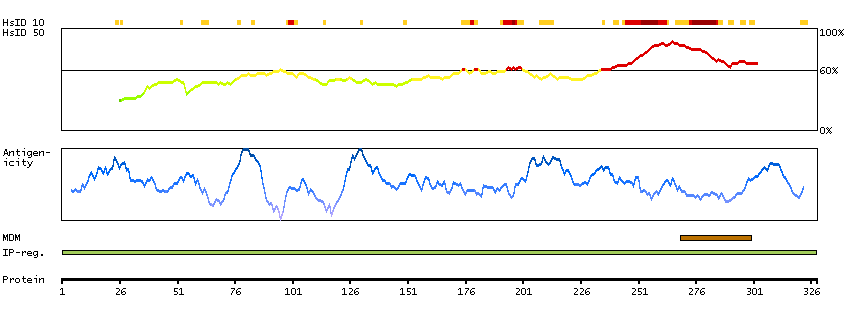
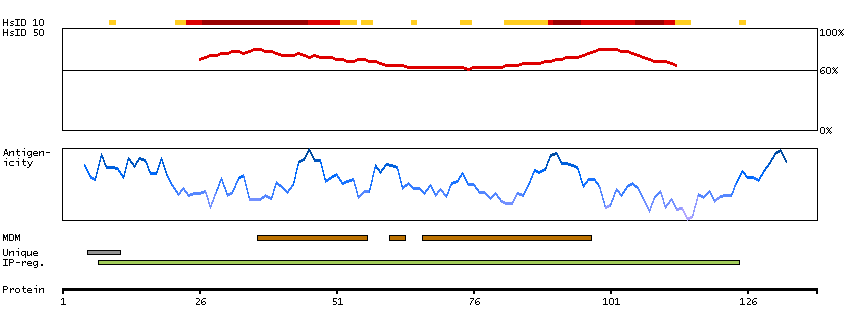
The protein encoded by this gene is a subunit of the vacuolar ATPase (v-ATPase), an heteromultimeric enzyme that is present in intracellular vesicles and in the plasma membrane of specialized cells, and which is essential for the acidification of diverse cellular components. V-ATPase is comprised of a membrane peripheral V(1) domain for ATP hydrolysis, and an integral membrane V(0) domain for proton translocation. The subunit encoded by this gene is a component of the V(0) domain. Mutations in this gene are a cause of both cutis laxa type II and wrinkly skin syndrome. [provided by RefSeq, Jul 2009]