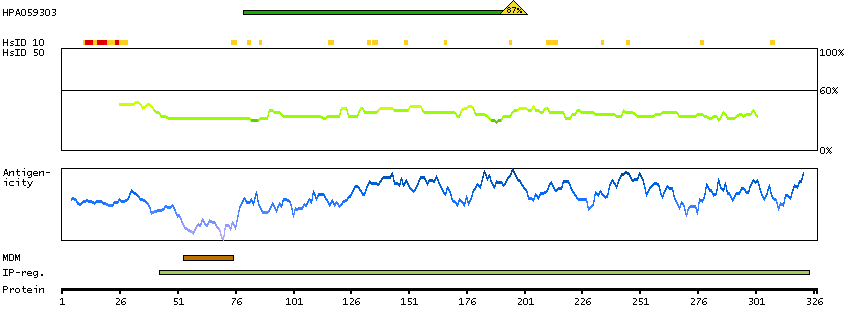
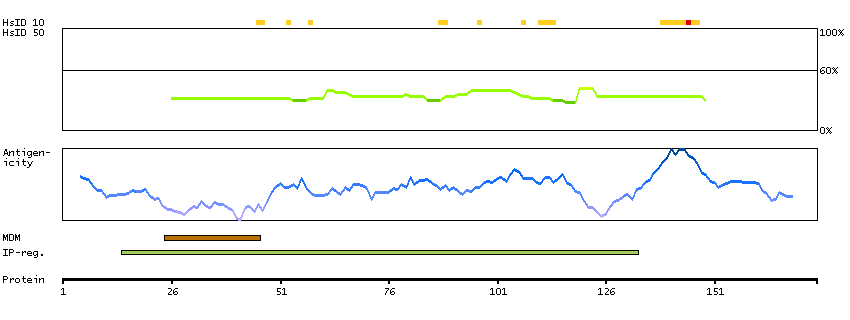
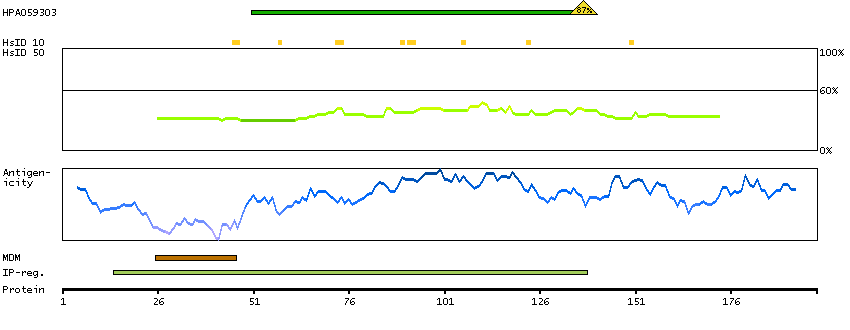
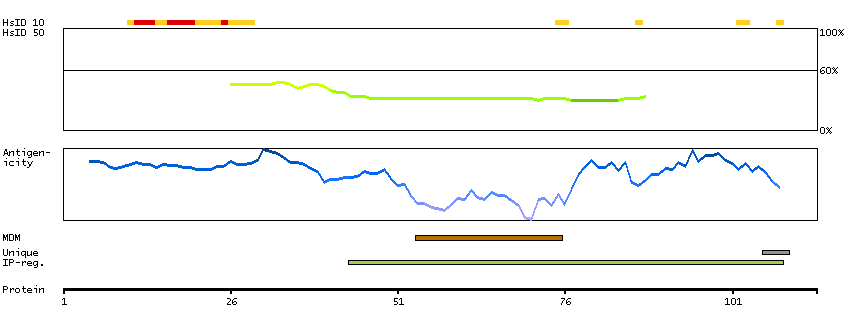
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
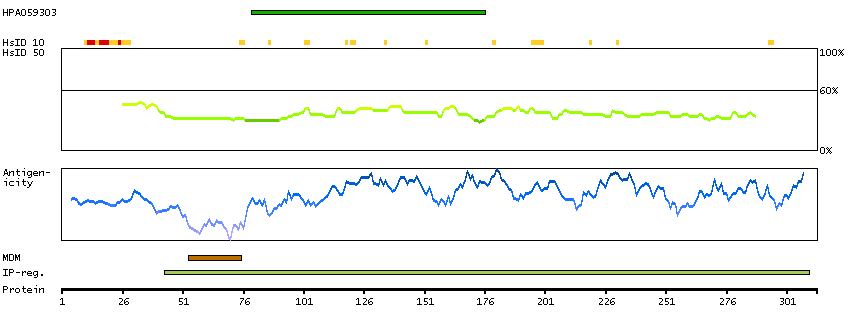
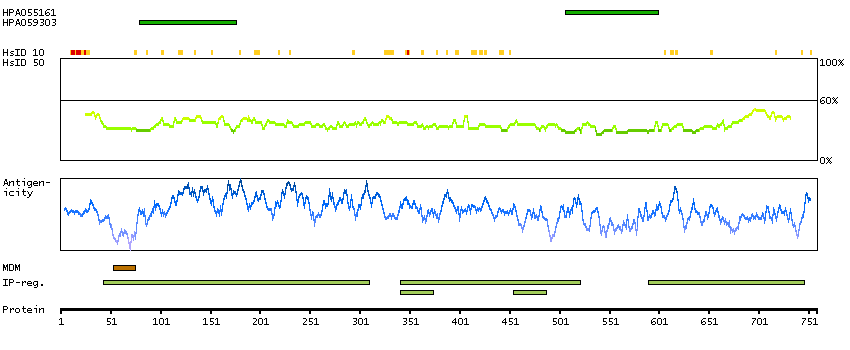
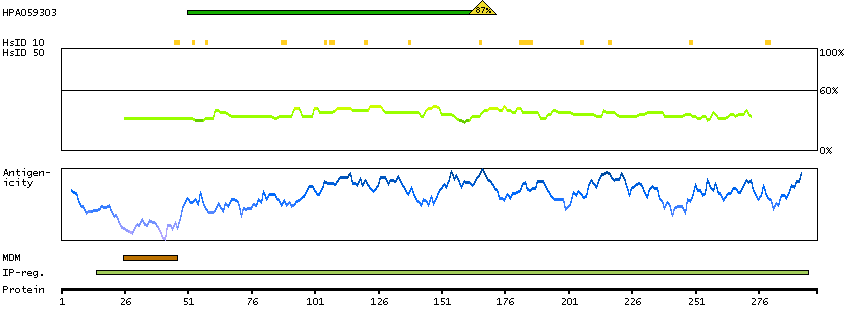
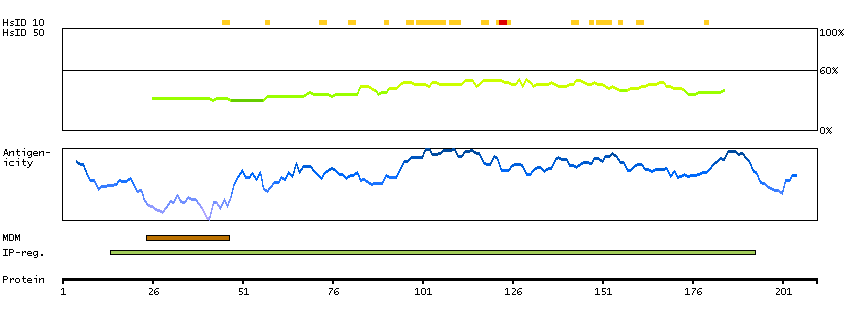
This gene is thought to play an important role in calcium homeostasis. The gene is expressed from two promoters and undergoes extensive alternative splicing. The encoded set of proteins share varying amounts of overlap near their N-termini but have substantial variations in their C-terminal domains resulting in distinct functional properties. The longest isoforms (a and f) include a C-terminal Aspartyl/Asparaginyl beta-hydroxylase domain that hydroxylates aspartic acid or asparagine residues in the epidermal growth factor (EGF)-like domains of some proteins, including protein C, coagulation factors VII, IX, and X, and the complement factors C1R and C1S. Other isoforms differ primarily in the C-terminal sequence and lack the hydroxylase domain, and some have been localized to the endoplasmic and sarcoplasmic reticulum. Some of these isoforms are found in complexes with calsequestrin, triadin, and the ryanodine receptor, and have been shown to regulate calcium release from the sarcoplasmic reticulum. Some isoforms have been implicated in metastasis. [provided by RefSeq, Sep 2009]