We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
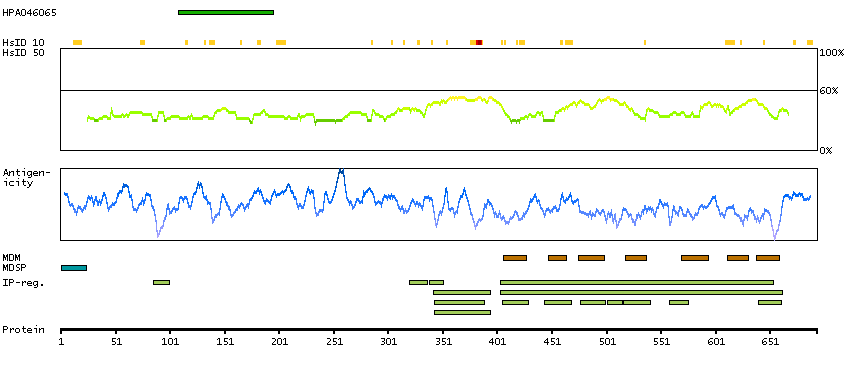
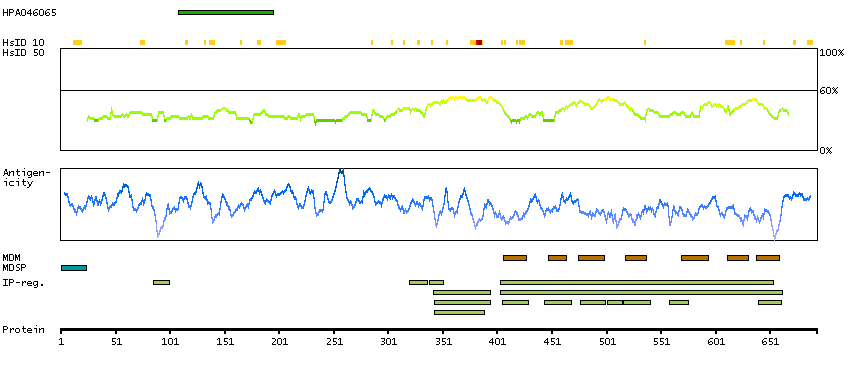
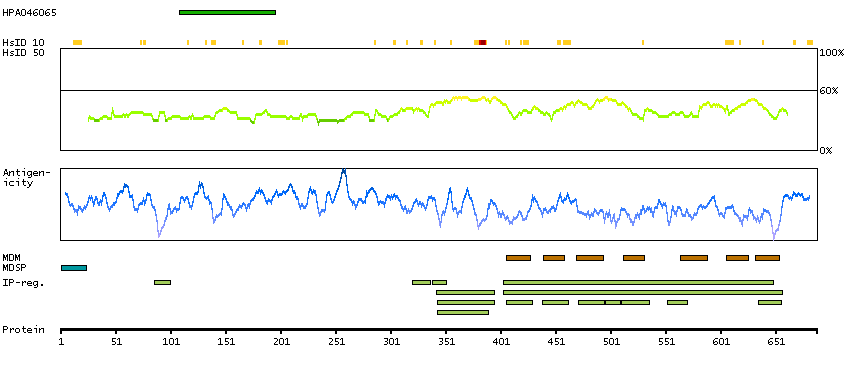
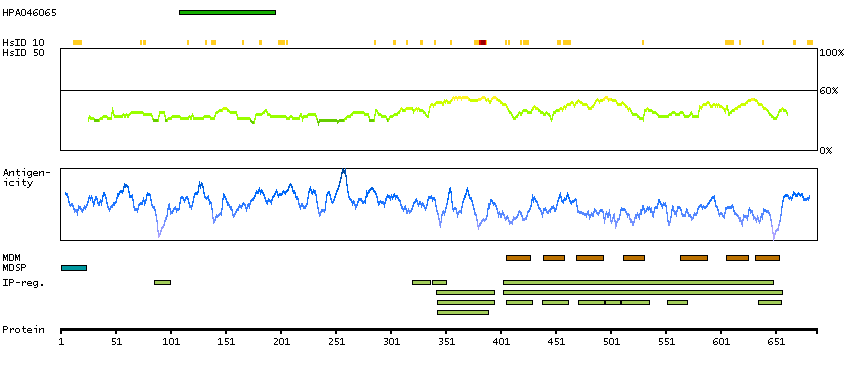
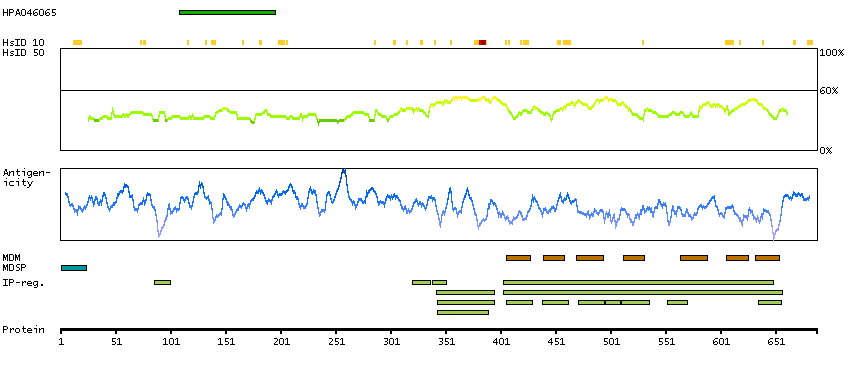
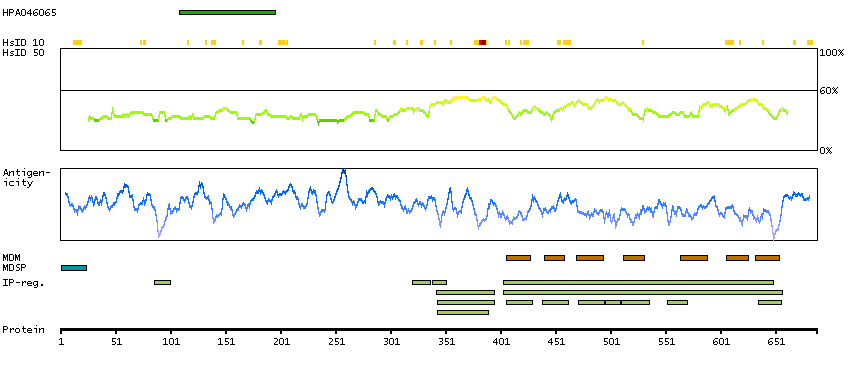
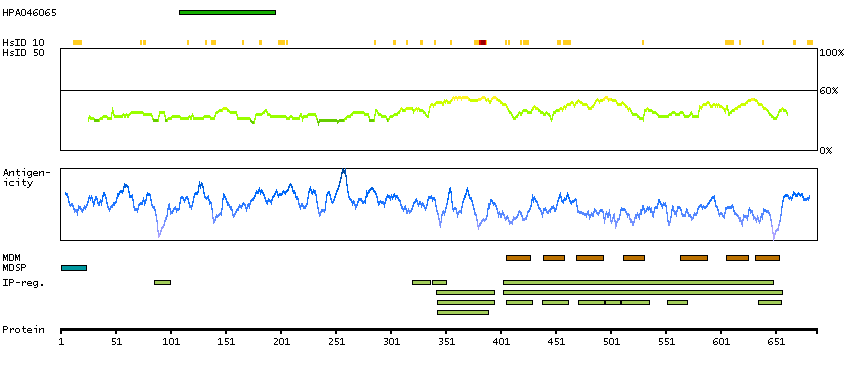
Adhesion G protein-coupled receptor G1 (HGNC Symbol)
Entrez gene summary
This gene encodes a member of the G protein-coupled receptor family and regulates brain cortical patterning. The encoded protein binds specifically to transglutaminase 2, a component of tissue and tumor stroma implicated as an inhibitor of tumor progression. Mutations in this gene are associated with a brain malformation known as bilateral frontoparietal polymicrogyria. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Feb 2014]