We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
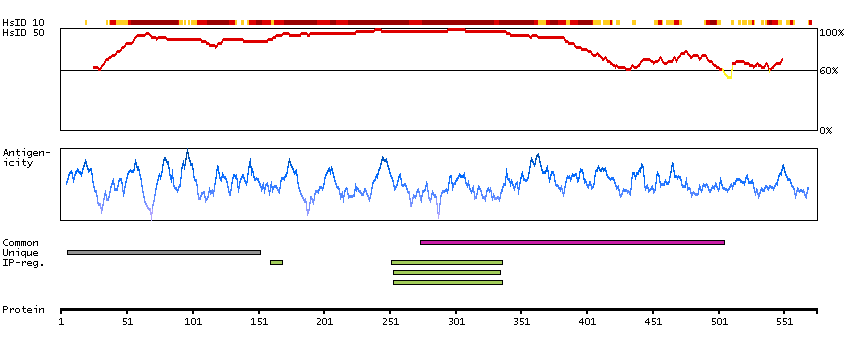
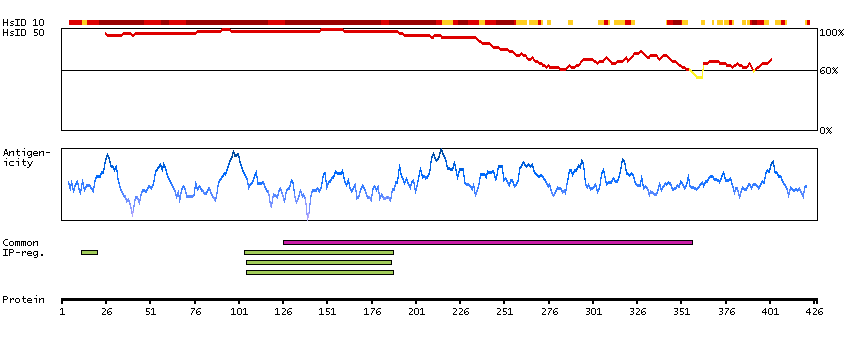
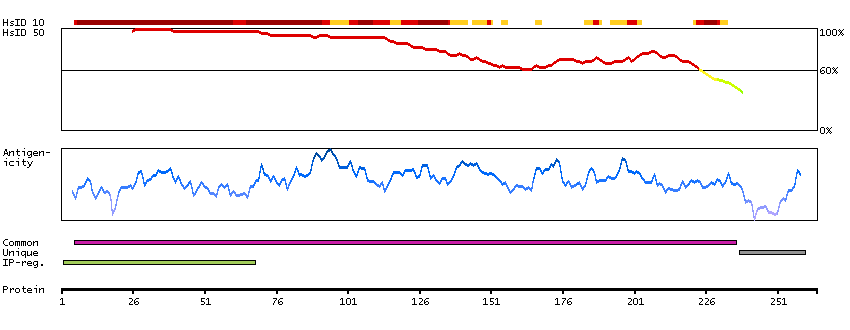
The protein encoded by this gene belongs to the COE (Collier/Olf/EBF) family of non-basic, helix-loop-helix transcription factors that have a well conserved DNA binding domain. The COE family proteins play an important role in variety of developmental processes. Studies in mouse suggest that this gene may be involved in the differentiation of osteoblasts. [provided by RefSeq, Oct 2011]