We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
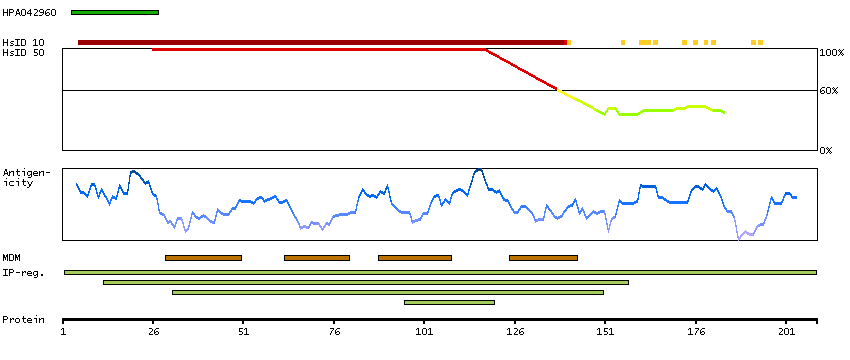
Estimation of protein expression could not be performed. View primary data.
DATA RELIABILITY
Data reliability description
RNA-based expert annotation gave inconclusive results. No RNA expression data detected and pending external verification. Caution, targets protein from more than one gene.