We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
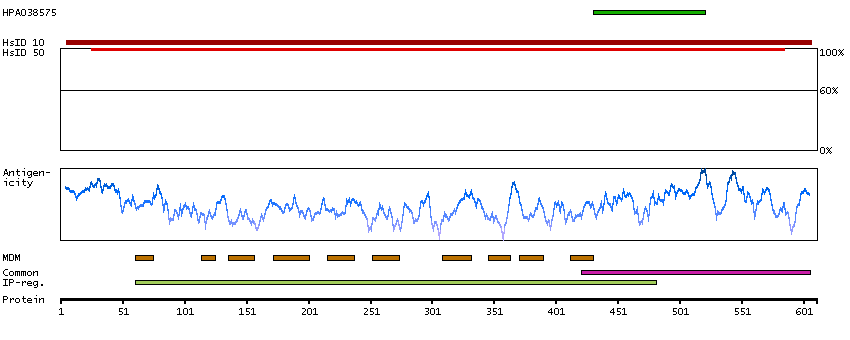
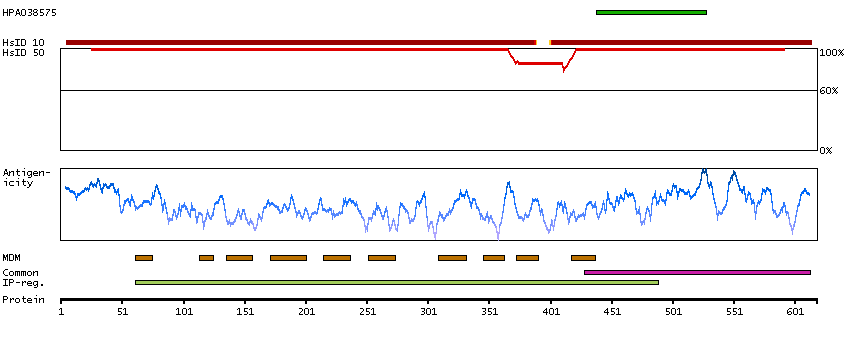
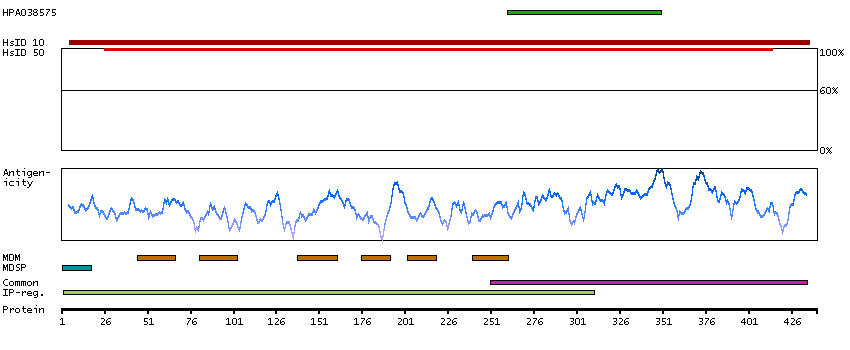
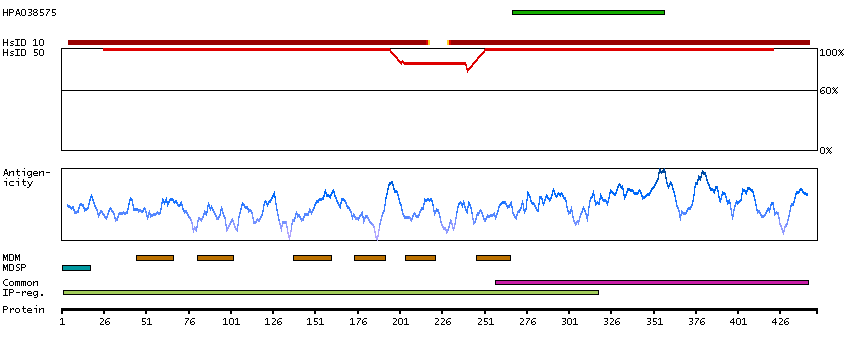
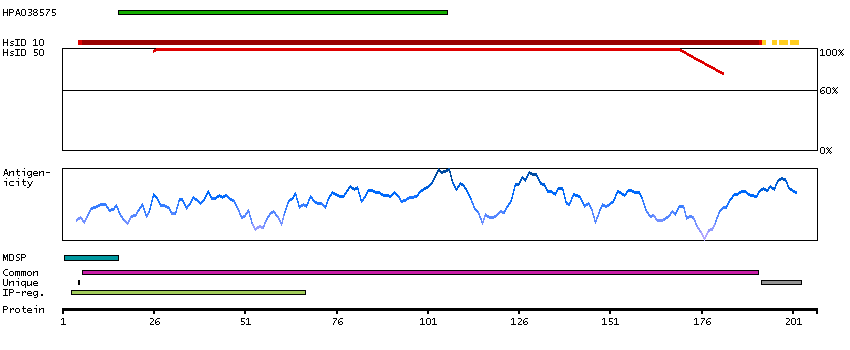
This gene encodes an alpha-1,2-mannosyltransferase enzyme that functions in lipid-linked oligosaccharide assembly. Mutations in this gene result in congenital disorder of glycosylation type Il. Multiple transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Dec 2008]