We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
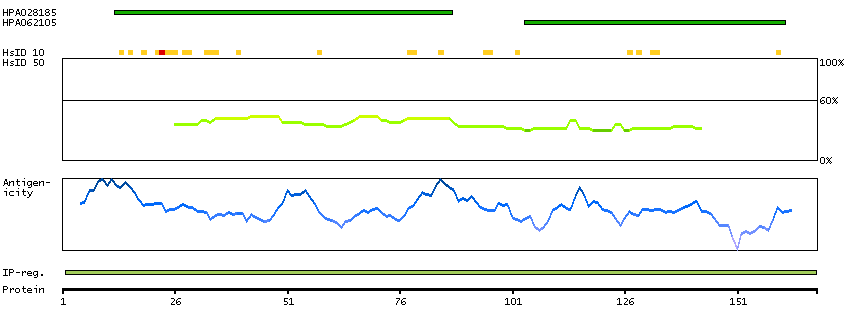
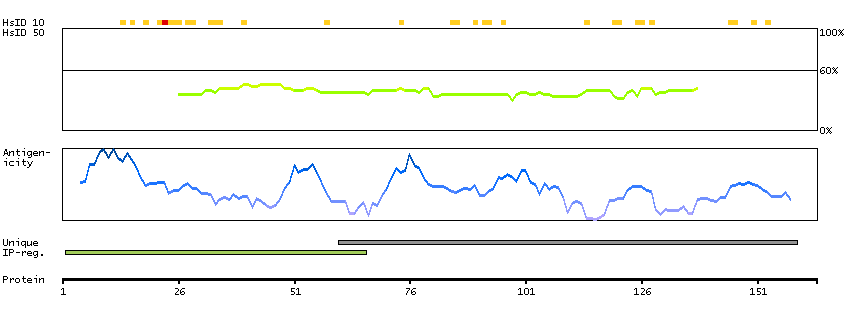
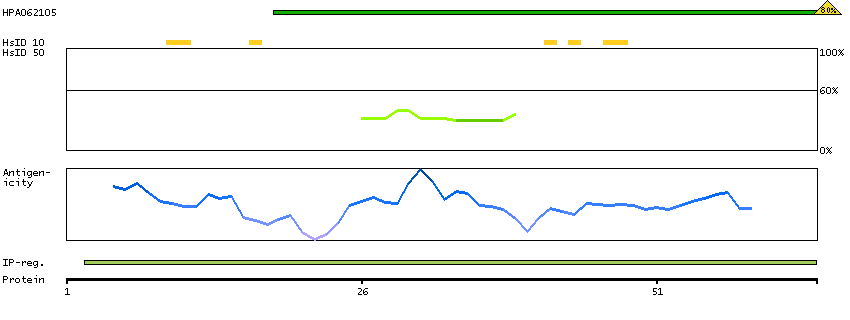
BCL2-associated agonist of cell death (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene is a member of the BCL-2 family. BCL-2 family members are known to be regulators of programmed cell death. This protein positively regulates cell apoptosis by forming heterodimers with BCL-xL and BCL-2, and reversing their death repressor activity. Proapoptotic activity of this protein is regulated through its phosphorylation. Protein kinases AKT and MAP kinase, as well as protein phosphatase calcineurin were found to be involved in the regulation of this protein. Alternative splicing of this gene results in two transcript variants which encode the same isoform. [provided by RefSeq, Jul 2008]
Q92934 [Direct mapping] Bcl2-associated agonist of cell death A0A024R562 [Target identity:100%; Query identity:100%] BCL2-antagonist of cell death, isoform CRA_c
Show all
Predicted intracellular proteins RAS pathway related proteins Cancer-related genes Candidate cancer biomarkers Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005515 [protein binding] GO:0005543 [phospholipid binding] GO:0005739 [mitochondrion] GO:0005741 [mitochondrial outer membrane] GO:0005829 [cytosol] GO:0006915 [apoptotic process] GO:0006919 [activation of cysteine-type endopeptidase activity involved in apoptotic process] GO:0007173 [epidermal growth factor receptor signaling pathway] GO:0008289 [lipid binding] GO:0008543 [fibroblast growth factor receptor signaling pathway] GO:0008656 [cysteine-type endopeptidase activator activity involved in apoptotic process] GO:0010508 [positive regulation of autophagy] GO:0010918 [positive regulation of mitochondrial membrane potential] GO:0012501 [programmed cell death] GO:0019901 [protein kinase binding] GO:0032024 [positive regulation of insulin secretion] GO:0033133 [positive regulation of glucokinase activity] GO:0038095 [Fc-epsilon receptor signaling pathway] GO:0042593 [glucose homeostasis] GO:0043065 [positive regulation of apoptotic process] GO:0043280 [positive regulation of cysteine-type endopeptidase activity involved in apoptotic process] GO:0044342 [type B pancreatic cell proliferation] GO:0045087 [innate immune response] GO:0045862 [positive regulation of proteolysis] GO:0045918 [negative regulation of cytolysis] GO:0046031 [ADP metabolic process] GO:0046034 [ATP metabolic process] GO:0046902 [regulation of mitochondrial membrane permeability] GO:0046931 [pore complex assembly] GO:0048011 [neurotrophin TRK receptor signaling pathway] GO:0048015 [phosphatidylinositol-mediated signaling] GO:0050679 [positive regulation of epithelial cell proliferation] GO:0071260 [cellular response to mechanical stimulus] GO:0071316 [cellular response to nicotine] GO:0071456 [cellular response to hypoxia] GO:0090200 [positive regulation of release of cytochrome c from mitochondria] GO:0097190 [apoptotic signaling pathway] GO:0097191 [extrinsic apoptotic signaling pathway] GO:0097193 [intrinsic apoptotic signaling pathway] GO:0097202 [activation of cysteine-type endopeptidase activity] GO:1900740 [positive regulation of protein insertion into mitochondrial membrane involved in apoptotic signaling pathway] GO:2000078 [positive regulation of type B pancreatic cell development] GO:2001244 [positive regulation of intrinsic apoptotic signaling pathway]
Q92934 [Direct mapping] Bcl2-associated agonist of cell death A0A024R562 [Target identity:100%; Query identity:100%] BCL2-antagonist of cell death, isoform CRA_c
Show all
Predicted intracellular proteins RAS pathway related proteins Cancer-related genes Candidate cancer biomarkers Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0001666 [response to hypoxia] GO:0001836 [release of cytochrome c from mitochondria] GO:0005515 [protein binding] GO:0005543 [phospholipid binding] GO:0005737 [cytoplasm] GO:0005739 [mitochondrion] GO:0005741 [mitochondrial outer membrane] GO:0005829 [cytosol] GO:0006007 [glucose catabolic process] GO:0006915 [apoptotic process] GO:0006919 [activation of cysteine-type endopeptidase activity involved in apoptotic process] GO:0007173 [epidermal growth factor receptor signaling pathway] GO:0007283 [spermatogenesis] GO:0008283 [cell proliferation] GO:0008289 [lipid binding] GO:0008543 [fibroblast growth factor receptor signaling pathway] GO:0008625 [extrinsic apoptotic signaling pathway via death domain receptors] GO:0008630 [intrinsic apoptotic signaling pathway in response to DNA damage] GO:0008656 [cysteine-type endopeptidase activator activity involved in apoptotic process] GO:0009725 [response to hormone] GO:0009749 [response to glucose] GO:0010033 [response to organic substance] GO:0010508 [positive regulation of autophagy] GO:0010918 [positive regulation of mitochondrial membrane potential] GO:0012501 [programmed cell death] GO:0014070 [response to organic cyclic compound] GO:0019050 [suppression by virus of host apoptotic process] GO:0019221 [cytokine-mediated signaling pathway] GO:0019901 [protein kinase binding] GO:0019903 [protein phosphatase binding] GO:0021987 [cerebral cortex development] GO:0030346 [protein phosphatase 2B binding] GO:0032024 [positive regulation of insulin secretion] GO:0032355 [response to estradiol] GO:0032570 [response to progesterone] GO:0033133 [positive regulation of glucokinase activity] GO:0033574 [response to testosterone] GO:0034201 [response to oleic acid] GO:0035774 [positive regulation of insulin secretion involved in cellular response to glucose stimulus] GO:0038095 [Fc-epsilon receptor signaling pathway] GO:0042493 [response to drug] GO:0042542 [response to hydrogen peroxide] GO:0042593 [glucose homeostasis] GO:0042981 [regulation of apoptotic process] GO:0043065 [positive regulation of apoptotic process] GO:0043200 [response to amino acid] GO:0043280 [positive regulation of cysteine-type endopeptidase activity involved in apoptotic process] GO:0043281 [regulation of cysteine-type endopeptidase activity involved in apoptotic process] GO:0043422 [protein kinase B binding] GO:0044342 [type B pancreatic cell proliferation] GO:0045087 [innate immune response] GO:0045471 [response to ethanol] GO:0045579 [positive regulation of B cell differentiation] GO:0045582 [positive regulation of T cell differentiation] GO:0045862 [positive regulation of proteolysis] GO:0045918 [negative regulation of cytolysis] GO:0046031 [ADP metabolic process] GO:0046034 [ATP metabolic process] GO:0046902 [regulation of mitochondrial membrane permeability] GO:0046931 [pore complex assembly] GO:0046982 [protein heterodimerization activity] GO:0048011 [neurotrophin TRK receptor signaling pathway] GO:0048015 [phosphatidylinositol-mediated signaling] GO:0050679 [positive regulation of epithelial cell proliferation] GO:0051384 [response to glucocorticoid] GO:0051592 [response to calcium ion] GO:0060139 [positive regulation of apoptotic process by virus] GO:0060154 [cellular process regulating host cell cycle in response to virus] GO:0071247 [cellular response to chromate] GO:0071260 [cellular response to mechanical stimulus] GO:0071316 [cellular response to nicotine] GO:0071396 [cellular response to lipid] GO:0071456 [cellular response to hypoxia] GO:0071889 [14-3-3 protein binding] GO:0090200 [positive regulation of release of cytochrome c from mitochondria] GO:0097190 [apoptotic signaling pathway] GO:0097191 [extrinsic apoptotic signaling pathway] GO:0097192 [extrinsic apoptotic signaling pathway in absence of ligand] GO:0097193 [intrinsic apoptotic signaling pathway] GO:0097202 [activation of cysteine-type endopeptidase activity] GO:1900740 [positive regulation of protein insertion into mitochondrial membrane involved in apoptotic signaling pathway] GO:1901216 [positive regulation of neuron death] GO:2000078 [positive regulation of type B pancreatic cell development] GO:2001244 [positive regulation of intrinsic apoptotic signaling pathway]