We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
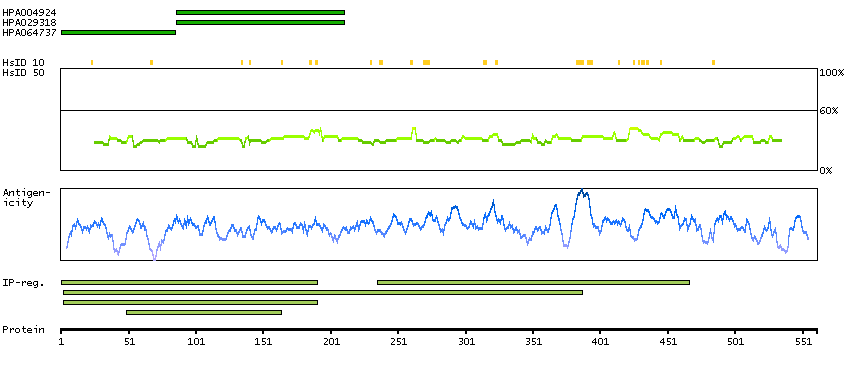
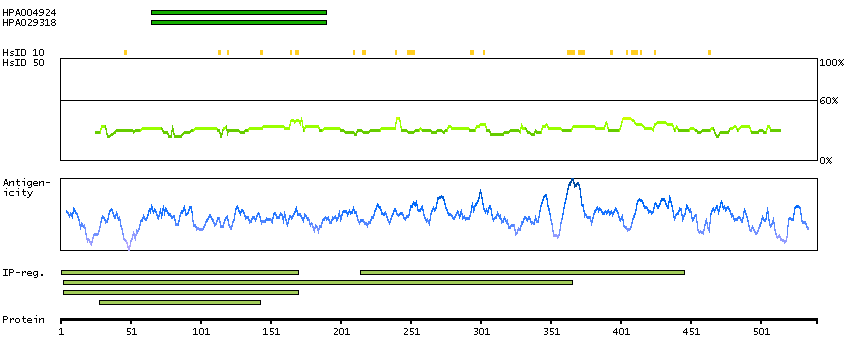
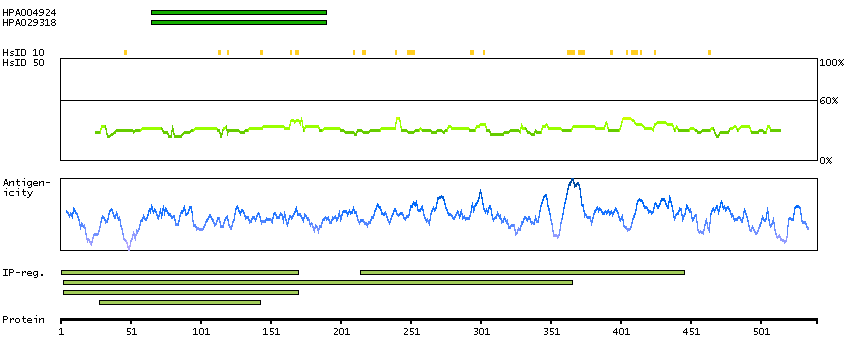
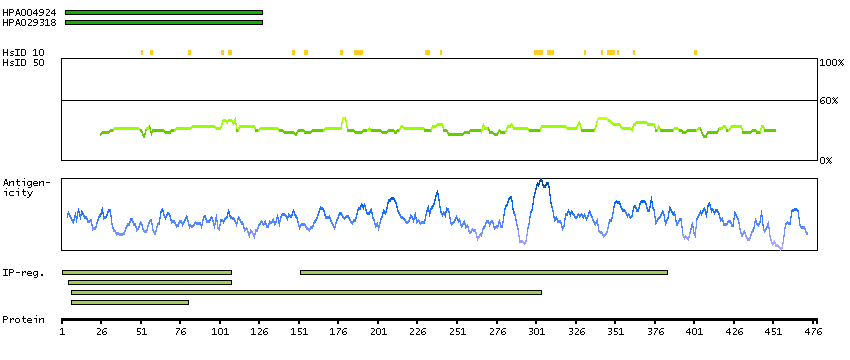
The protein encoded by this gene is involved in the synthesis of asparagine. This gene complements a mutation in the temperature-sensitive hamster mutant ts11, which blocks progression through the G1 phase of the cell cycle at nonpermissive temperature. Alternatively spliced transcript variants have been described for this gene. [provided by RefSeq, May 2010]
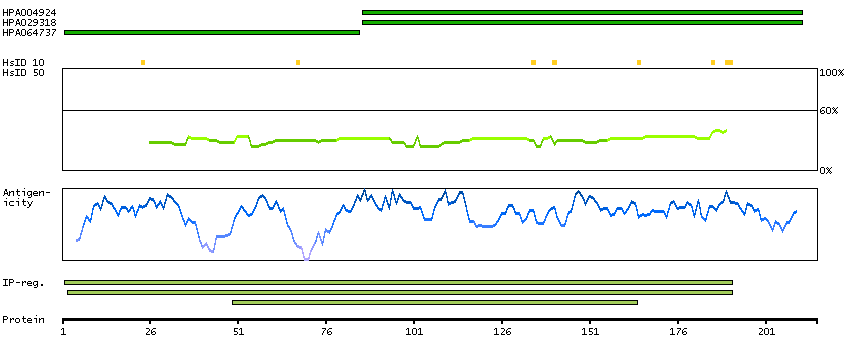
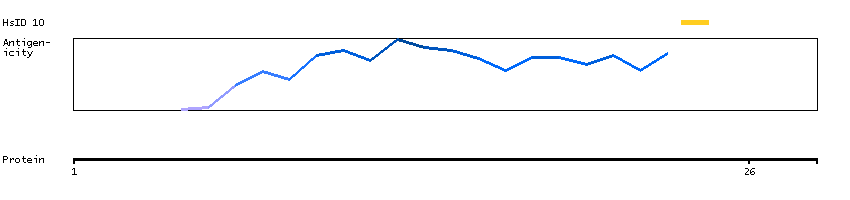
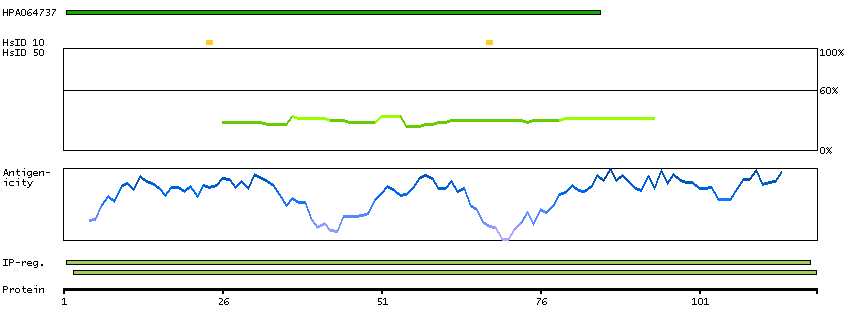
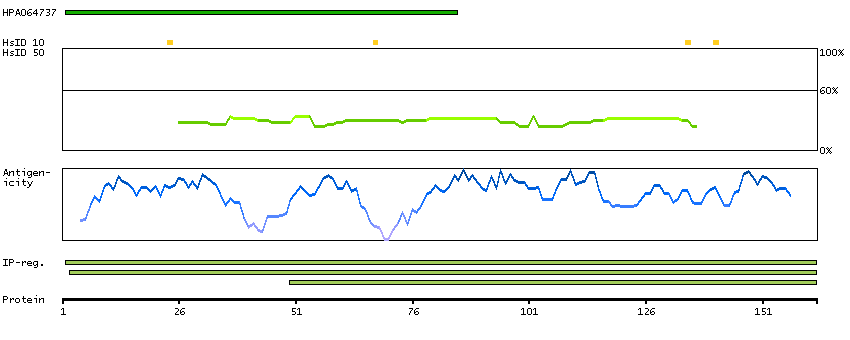
Enzymes ENZYME proteins Ligase Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Enzymes ENZYME proteins Ligase Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Enzymes ENZYME proteins Ligase Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Enzymes ENZYME proteins Ligase Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Enzymes ENZYME proteins Ligase Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Enzymes ENZYME proteins Ligase Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0001889 [liver development] GO:0004066 [asparagine synthase (glutamine-hydrolyzing) activity] GO:0005515 [protein binding] GO:0005524 [ATP binding] GO:0005829 [cytosol] GO:0006520 [cellular amino acid metabolic process] GO:0006529 [asparagine biosynthetic process] GO:0006541 [glutamine metabolic process] GO:0008652 [cellular amino acid biosynthetic process] GO:0009416 [response to light stimulus] GO:0009612 [response to mechanical stimulus] GO:0009636 [response to toxic substance] GO:0030968 [endoplasmic reticulum unfolded protein response] GO:0031427 [response to methotrexate] GO:0031667 [response to nutrient levels] GO:0032354 [response to follicle-stimulating hormone] GO:0032870 [cellular response to hormone stimulus] GO:0034641 [cellular nitrogen compound metabolic process] GO:0036499 [PERK-mediated unfolded protein response] GO:0042149 [cellular response to glucose starvation] GO:0042803 [protein homodimerization activity] GO:0043066 [negative regulation of apoptotic process] GO:0043200 [response to amino acid] GO:0044267 [cellular protein metabolic process] GO:0044281 [small molecule metabolic process] GO:0045931 [positive regulation of mitotic cell cycle] GO:0048037 [cofactor binding] GO:0070981 [L-asparagine biosynthetic process]