We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Protein kinase C and casein kinase substrate in neurons 2 (HGNC Symbol)
Entrez gene summary
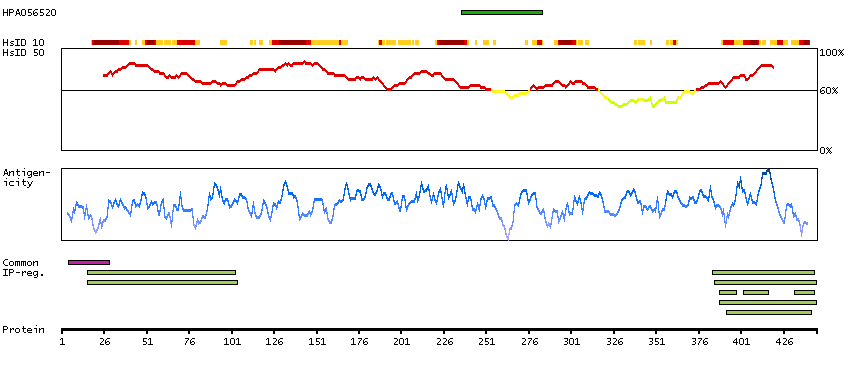
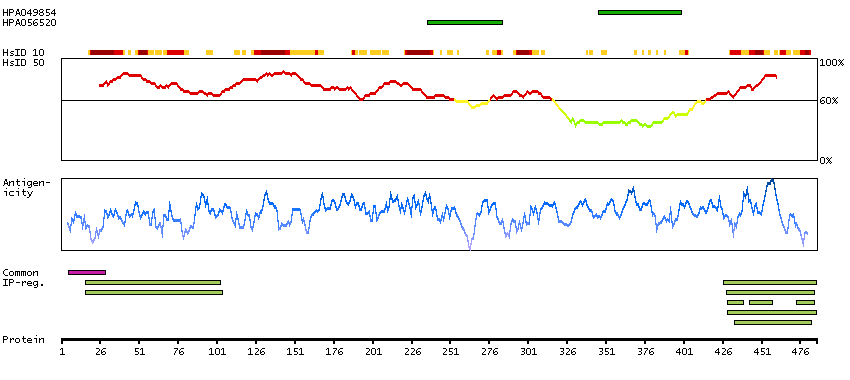
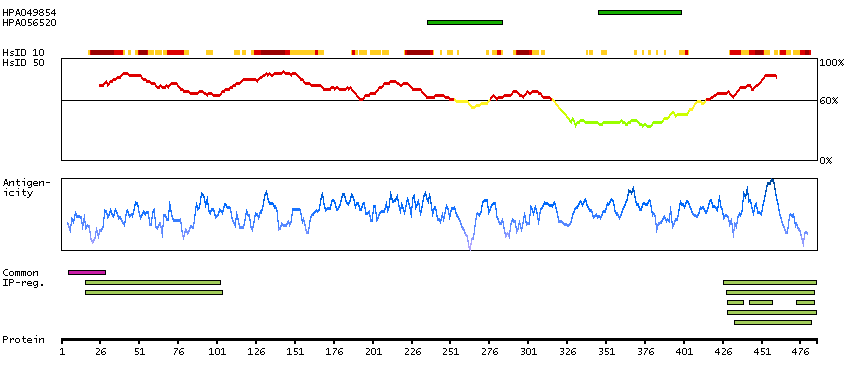
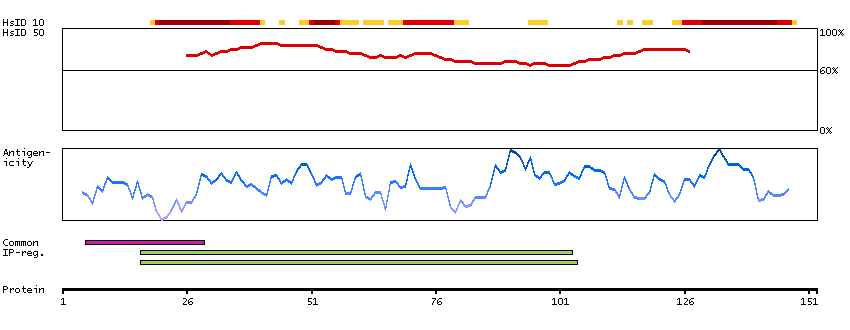
This gene is a member of the protein kinase C and casein kinase substrate in neurons family. The encoded protein is involved in linking the actin cytoskeleton with vesicle formation by regulating tubulin polymerization. Alternative splicing results in multiple transcript variants. [provided by RefSeq, May 2010]
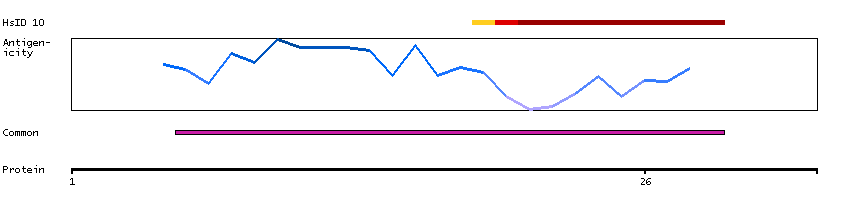
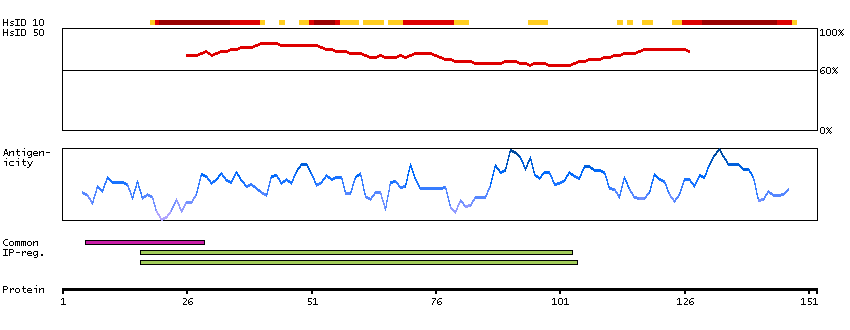
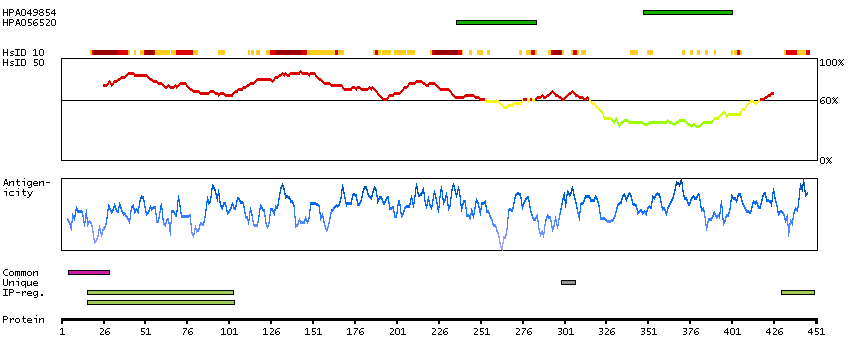
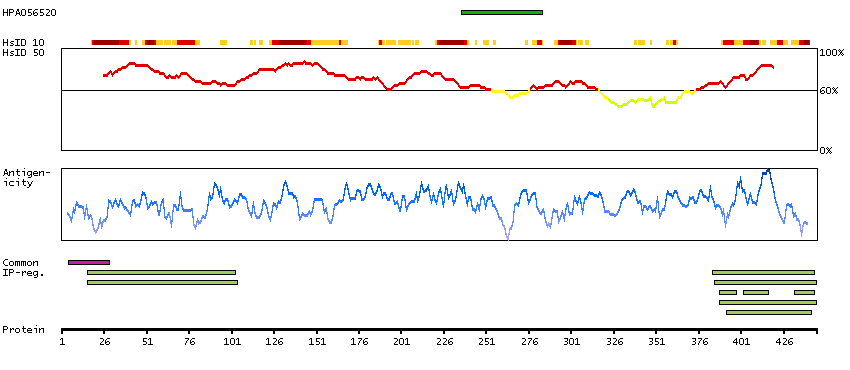
Q9UNF0 [Direct mapping] Protein kinase C and casein kinase substrate in neurons protein 2 Q6FIA3 [Target identity:100%; Query identity:100%] PACSIN2 protein
Show all
Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Q9UNF0 [Direct mapping] Protein kinase C and casein kinase substrate in neurons protein 2 Q6FIA3 [Target identity:100%; Query identity:100%] PACSIN2 protein
Show all
Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)